Reductive elimination is an elementary step in organometallic chemistry in which the oxidation state of the metal center decreases while forming a new covalent bond between two ligands. It is the microscopic reverse of oxidative addition, and is often the product-forming step in many catalytic processes. Since oxidative addition and reductive elimination are reverse reactions, the same mechanisms apply for both processes, and the product equilibrium depends on the thermodynamics of both directions.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.
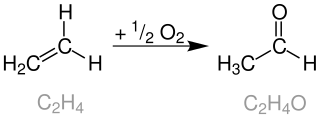
The Wacker process or the Hoechst-Wacker process refers to the oxidation of ethylene to acetaldehyde in the presence of palladium(II) chloride as the catalyst. This chemical reaction was one of the first homogeneous catalysis with organopalladium chemistry applied on an industrial scale.
Carbon–hydrogen bond functionalization is a type of reaction in which a carbon–hydrogen bond is cleaved and replaced with a carbon–X bond. The term usually implies that a transition metal is involved in the C-H cleavage process. Reactions classified by the term typically involve the hydrocarbon first to react with a metal catalyst to create an organometallic complex in which the hydrocarbon is coordinated to the inner-sphere of a metal, either via an intermediate "alkane or arene complex" or as a transition state leading to a "M−C" intermediate. The intermediate of this first step can then undergo subsequent reactions to produce the functionalized product. Important to this definition is the requirement that during the C–H cleavage event, the hydrocarbyl species remains associated in the inner-sphere and under the influence of "M".
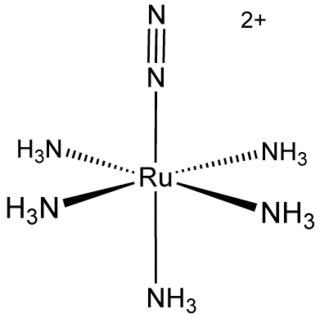
Transition metal dinitrogen complexes are coordination compounds that contain transition metals as ion centers the dinitrogen molecules (N2) as ligands.

The Liebeskind–Srogl coupling reaction is an organic reaction forming a new carbon–carbon bond from a thioester and a boronic acid using a metal catalyst. It is a cross-coupling reaction. This reaction was invented by and named after Jiri Srogl from the Academy of Sciences, Czech Republic, and Lanny S. Liebeskind from Emory University, Atlanta, Georgia, USA. There are three generations of this reaction, with the first generation shown below. The original transformation used catalytic Pd(0), TFP = tris(2-furyl)phosphine as an additional ligand and stoichiometric CuTC = copper(I) thiophene-2-carboxylate as a co-metal catalyst. The overall reaction scheme is shown below.
Jay Kazuo Kochi (1927–2008) was an American physical organometallic chemist who held lectureship at Harvard University, and faculty positions at Case Institute of Technology, 1962-1969,, Indiana University, 1969 to 1984, and the University of Houston, 1984 to 2008.
Metal carbon dioxide complexes are coordination complexes that contain carbon dioxide ligands. Aside from the fundamental interest in the coordination chemistry of simple molecules, studies in this field are motivated by the possibility that transition metals might catalyze useful transformations of CO2. This research is relevant both to organic synthesis and to the production of "solar fuels" that would avoid the use of petroleum-based fuels.

The Catellani reaction was discovered by Marta Catellani and co-workers in 1997. The reaction uses aryl iodides to perform bi- or tri-functionalization, including C-H functionalization of the unsubstituted ortho position(s), followed a terminating cross-coupling reaction at the ipso position. This cross-coupling cascade reaction depends on the ortho-directing transient mediator, norbornene.

Transition-metal allyl complexes are coordination complexes with allyl and its derivatives as ligands. Allyl is the radical with the connectivity CH2CHCH2, although as a ligand it is usually viewed as an allyl anion CH2=CH−CH2−, which is usually described as two equivalent resonance structures.
Karen Ila Goldberg is an American chemist, currently the Vagelos Professor of Energy Research at University of Pennsylvania. Goldberg is most known for her work in inorganic and organometallic chemistry. Her most recent research focuses on catalysis, particularly on developing catalysts for oxidation, as well as the synthesis and activation of molecular oxygen. In 2018, Goldberg was elected to the National Academy of Sciences.
In organic chemistry, the Fujiwara–Moritani reaction is a type of cross coupling reaction where an aromatic C-H bond is directly coupled to an olefinic C-H bond, generating a new C-C bond. This reaction is performed in the presence of a transition metal, typically palladium. The reaction was discovered by Yuzo Fujiwara and Ichiro Moritani in 1967. An external oxidant is required to this reaction to be run catalytically. Thus, this reaction can be classified as a C-H activation reaction, an oxidative Heck reaction, and a C-H olefination. Surprisingly, the Fujiwara–Moritani reaction was discovered before the Heck reaction.
In organic and organometallic chemistry, dialkylbiaryl phosphine (or dialkylbiarylphosphine) ligands are phosphorus-containing supporting ligands that are used to modulate the chemical reactivity of palladium and other transition metal based catalysts. They were first described by Stephen L. Buchwald in 1998 for applications in palladium-catalyzed coupling reactions to form carbon-nitrogen and carbon-carbon bonds. Before their development, use of first- or second-generation phosphine ligands for palladium-catalyzed C-N bond-forming cross-coupling (e.g., tris(o-tolyl)phosphine and BINAP, respectively) necessitated harsh conditions, and the scope of the transformation was severely limited. The Suzuki-Miyaura and Negishi cross-coupling reactions were typically performed with Pd(PPh3)4 as catalyst and were mostly limited to aryl bromides and iodides at elevated temperatures, while the widely available aryl chlorides were unreactive. The development of new classes of ligands was needed to address these limitations.
Cobalt(II)–porphyrin catalysis is a process in which a Co(II) porphyrin complex acts as a catalyst, inducing and accelerating a chemical reaction.
The Mukaiyama hydration is an organic reaction involving formal addition of an equivalent of water across an olefin by the action of catalytic bis(acetylacetonato)cobalt(II) complex, phenylsilane and atmospheric oxygen to produce an alcohol with Markovnikov selectivity.

In organometallic chemistry, the activation of cyclopropanes by transition metals is a research theme with implications for organic synthesis and homogeneous catalysis. Being highly strained, cyclopropanes are prone to oxidative addition to transition metal complexes. The resulting metallacycles are susceptible to a variety of reactions. These reactions are rare examples of C-C bond activation. The rarity of C-C activation processes has been attributed to Steric effects that protect C-C bonds. Furthermore, the directionality of C-C bonds as compared to C-H bonds makes orbital interaction with transition metals less favorable. Thermodynamically, C-C bond activation is more favored than C-H bond activation as the strength of a typical C-C bond is around 90 kcal per mole while the strength of a typical unactivated C-H bond is around 104 kcal per mole.
Organoniobium chemistry is the chemistry of compounds containing niobium-carbon (Nb-C) bonds. Compared to the other group 5 transition metal organometallics, the chemistry of organoniobium compounds most closely resembles that of organotantalum compounds. Organoniobium compounds of oxidation states +5, +4, +3, +2, +1, 0, -1, and -3 have been prepared, with the +5 oxidation state being the most common.

Phosphenium ions, not to be confused with phosphonium or phosphirenium, are divalent cations of phosphorus of the form [PR2]+. Phosphenium ions have long been proposed as reaction intermediates.

The Mizoroki−Heck coupling of aryl halides and alkenes to form C(sp2)–C(sp2) bonds has become a staple transformation in organic synthesis, owing to its broad functional group compatibility and varied scope. In stark contrast, the palladium-catalyzed reductive Heck reaction has received considerably less attention, despite the fact that early reports of this reaction date back almost half a century. From the perspective of retrosynthetic logic, this transformation is highly enabling because it can forge alkyl–aryl linkages from widely available alkenes, rather than from the less accessible and/or more expensive alkyl halide or organometallic C(sp3) synthons that are needed in a classical aryl/alkyl cross-coupling.
β-carbon elimination is a type of reaction in organometallic chemistry wherein an allyl ligand bonded to a metal center is broken into the corresponding metal-bonded alkyl (aryl) ligand and an alkene. It is a subgroup of elimination reactions. Though less common and less understood than β-hydride elimination, it is an important step involved in some olefin polymerization processes and transition-metal-catalyzed organic reactions.







