| Crouzonodermoskeletal syndrome | |
|---|---|
| Other names | Crouzon syndrome with acanthosis nigricans |
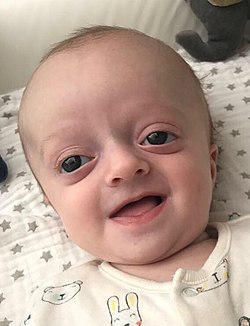
Crouzonodermoskeletal syndrome is a disorder characterized by the premature joining of certain bones of the skull (craniosynostosis) during development and a skin condition called acanthosis nigricans. [1]
Contents
Some of the signs and symptoms of Crouzonodermoskeletal syndrome are similar to those seen with Crouzon syndrome. They include prematurely fused skull bones, which affect the shape of the head and face; wide-set, bulging eyes due to shallow eye sockets; eyes that do not point in the same direction (strabismus); a small, beaked nose; and an underdeveloped upper jaw. People with these conditions are generally of normal intelligence. [2]
Several features distinguish Crouzonodermoskeletal syndrome from Crouzon syndrome. People with Crouzonodermoskeletal syndrome have acanthosis nigricans, a skin condition characterized by thick, dark, velvety skin in body folds and creases, including the neck and underarms. In addition, subtle changes may be seen in the bones of the spine (vertebrae). Noncancerous growths called cementomas may develop in the jaw during young adulthood. Crouzonodermoskeletal syndrome is rare; the condition is seen in about 1 per million people.[ citation needed ]