Related Research Articles
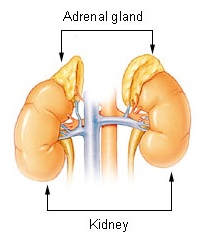
Adrenal insufficiency is a condition in which the adrenal glands do not produce adequate amounts of steroid hormones. The adrenal glands—also referred to as the adrenal cortex—normally secrete glucocorticoids, mineralocorticoids, and androgens. These hormones are important in regulating blood pressure, electrolytes, and metabolism as a whole. Deficiency of these hormones leads to symptoms ranging from abdominal pain, vomiting, muscle weakness and fatigue, low blood pressure, depression, mood and personality changes to organ failure and shock. Adrenal crisis may occur if a person having adrenal insufficiency experiences stresses, such as an accident, injury, surgery, or severe infection; this is a life-threatening medical condition resulting from severe deficiency of cortisol in the body. Death may quickly follow.

Growth hormone deficiency (GHD), or human growth hormone deficiency, is a medical condition resulting from not enough growth hormone (GH). Generally the most noticeable symptom is that an individual attains a short height. Newborns may also present low blood sugar or a small penis size. In adults there may be decreased muscle mass, high cholesterol levels, or poor bone density.
Immunodeficiency, also known as immunocompromisation, is a state in which the immune system's ability to fight infectious diseases and cancer is compromised or entirely absent. Most cases are acquired ("secondary") due to extrinsic factors that affect the patient's immune system. Examples of these extrinsic factors include HIV infection and environmental factors, such as nutrition. Immunocompromisation may also be due to genetic diseases/flaws such as SCID.

Pituitary adenomas are tumors that occur in the pituitary gland. Most pituitary tumors are benign, approximately 35% are invasive and just 0.1% to 0.2% are carcinomas. Pituitary adenomas represent from 10% to 25% of all intracranial neoplasms, with an estimated prevalence rate in the general population of approximately 17%.

X-linked agammaglobulinemia (XLA) is a rare genetic disorder discovered in 1952 that affects the body's ability to fight infection. As the form of agammaglobulinemia that is X-linked, it is much more common in males. In people with XLA, the white blood cell formation process does not generate mature B cells, which manifests as a complete or near-complete lack of proteins called gamma globulins, including antibodies, in their bloodstream. B cells are part of the immune system and normally manufacture antibodies, which defend the body from infections by sustaining a humoral immunity response. Patients with untreated XLA are prone to develop serious and even fatal infections. A mutation occurs at the Bruton's tyrosine kinase (Btk) gene that leads to a severe block in B cell development and a reduced immunoglobulin production in the serum. Btk is particularly responsible for mediating B cell development and maturation through a signaling effect on the B cell receptor BCR. Patients typically present in early childhood with recurrent infections, in particular with extracellular, encapsulated bacteria. XLA is deemed to have a relatively low incidence of disease, with an occurrence rate of approximately 1 in 200,000 live births and a frequency of about 1 in 100,000 male newborns. It has no ethnic predisposition. XLA is treated by infusion of human antibody. Treatment with pooled gamma globulin cannot restore a functional population of B cells, but it is sufficient to reduce the severity and number of infections due to the passive immunity granted by the exogenous antibodies.
Hypogammaglobulinemia is an immune system disorder in which not enough gamma globulins are produced in the blood. This results in a lower antibody count, which impairs the immune system, increasing risk of infection. Hypogammaglobulinemia may result from a variety of primary genetic immune system defects, such as common variable immunodeficiency, or it may be caused by secondary effects such as medication, blood cancer, or poor nutrition, or loss of gamma globulins in urine, as in nonselective glomerular proteinuria. Patients with hypogammaglobulinemia have reduced immune function; important considerations include avoiding use of live vaccines, and take precautionary measures when traveling to regions with endemic disease or poor sanitation such as receiving immunizations, taking antibiotics abroad, drinking only safe or boiled water, arranging appropriate medical cover in advance of travel, and ensuring continuation of any immunoglobulin infusions needed.
Common variable immunodeficiency (CVID) is an inborn immune disorder characterized by recurrent infections and low antibody levels, specifically in immunoglobulin (Ig) types IgG, IgM, and IgA. Symptoms generally include high susceptibility to pathogens, chronic lung disease, as well as inflammation and infection of the gastrointestinal tract.

Adrenocorticotropic hormone deficiency is a rare disorder characterized by secondary adrenal insufficiency with minimal or no cortisol production and normal pituitary hormone secretion apart from ACTH. ACTH deficiency may be congenital or acquired, and its symptoms are clinically similar to those of glucocorticoid deficiency. Symptoms consist of weight loss, diminished appetite, muscle weakness, nausea, vomiting, and hypotension. Low blood sugar and hyponatremia are possible; however, blood potassium levels typically remain normal because affected patients are deficient in glucocorticoids rather than mineralocorticoids because of their intact renin-angiotensin-aldosterone system. ACTH may be undetectable in blood tests, and cortisol is abnormally low. Glucocorticoid replacement therapy is required. With the exception of stressful situations, some patients with mild or nearly asymptomatic disease may not require glucocorticoid replacement therapy. As of 2008 about two hundred cases have been described in the literature.
Hyper IgM syndrome is a rare primary immune deficiency disorders characterized by low or absent levels of serum IgG, IgA, IgE and normal or increased levels of serum IgM.

Endocrine diseases are disorders of the endocrine system. The branch of medicine associated with endocrine disorders is known as endocrinology.
Laron syndrome (LS), also known as growth hormone insensitivity or growth hormone receptor deficiency (GHRD), is an autosomal recessive disorder characterized by a lack of insulin-like growth factor 1 production in response to growth hormone. It is usually caused by inherited growth hormone receptor (GHR) mutations.

WHIM syndrome is a rare congenital immunodeficiency disorder characterized by chronic noncyclic neutropenia.

Nuclear factor NF-kappa-B p100 subunit is a protein that in humans is encoded by the NFKB2 gene.

Immunoglobulin superfamily, member 1 is a plasma membrane glycoprotein encoded by the IGSF1 gene, which maps to the X chromosome in humans and other mammalian species.

IgG deficiency is a form of dysgammaglobulinemia where the proportional levels of the IgG isotype are reduced relative to other immunoglobulin isotypes.
Gonadotropin-releasing hormone (GnRH) insensitivity also known as Isolated gonadotropin-releasing hormone (GnRH)deficiency (IGD) is a rare autosomal recessive genetic and endocrine syndrome which is characterized by inactivating mutations of the gonadotropin-releasing hormone receptor (GnRHR) and thus an insensitivity of the receptor to gonadotropin-releasing hormone (GnRH), resulting in a partial or complete loss of the ability of the gonads to synthesize the sex hormones. The condition manifests itself as isolated hypogonadotropic hypogonadism (IHH), presenting with symptoms such as delayed, reduced, or absent puberty, low or complete lack of libido, and infertility, and is the predominant cause of IHH when it does not present alongside anosmia.
Hypogonadotropic hypogonadism (HH), is due to problems with either the hypothalamus or pituitary gland affecting the hypothalamic-pituitary-gonadal axis. Hypothalamic disorders result from a deficiency in the release of gonadotropic releasing hormone (GnRH), while pituitary gland disorders are due to a deficiency in the release of gonadotropins from the anterior pituitary. GnRH is the central regulator in reproductive function and sexual development via the HPG axis. GnRH is released by GnRH neurons, which are hypothalamic neuroendocrine cells, into the hypophyseal portal system acting on gonadotrophs in the anterior pituitary. The release of gonadotropins, LH and FSH, act on the gonads for the development and maintenance of proper adult reproductive physiology. LH acts on Leydig cells in the male testes and theca cells in the female. FSH acts on Sertoli cells in the male and follicular cells in the female. Combined this causes the secretion of gonadal sex steroids and the initiation of folliculogenesis and spermatogenesis. The production of sex steroids forms a negative feedback loop acting on both the anterior pituitary and hypothalamus causing a pulsatile secretion of GnRH. GnRH neurons lack sex steroid receptors and mediators such as kisspeptin stimulate GnRH neurons for pulsatile secretion of GnRH.
Cortisone reductase deficiency is caused by dysregulation of the 11β-hydroxysteroid dehydrogenase type 1 enzyme (11β-HSD1), otherwise known as cortisone reductase, a bi-directional enzyme, which catalyzes the interconversion of cortisone to cortisol in the presence of NADH as a co-factor. If levels of NADH are low, the enzyme catalyses the reverse reaction, from cortisol to cortisone, using NAD+ as a co-factor.
Cortisol is a glucocorticoid that plays a variety of roles in many different biochemical pathways, including, but not limited to: gluconeogenesis, suppressing immune system responses and carbohydrate metabolism.
One of the symptoms of cortisone reductase deficiency is hyperandrogenism, resulting from activation of the Hypothalamic–pituitary–adrenal axis. The deficiency has been known to exhibit symptoms of other disorders such as Polycystic Ovary Syndrome in women. Cortisone Reductase Deficiency alone has been reported in fewer than ten cases in total, all but one case were women. Elevated activity of 11β-HSD1 can lead to obesity or Type II Diabetes, because of the role of cortisol in carbohydrate metabolism and gluconeogenesis.
Nuclear factor-kappa B Essential Modulator (NEMO) deficiency syndrome is a rare type of primary immunodeficiency disease that has a highly variable set of symptoms and prognoses. It mainly affects the skin and immune system but has the potential to affect all parts of the body, including the lungs, urinary tract and gastrointestinal tract. It is a monogenetic disease caused by mutation in the IKBKG gene. NEMO is the modulator protein in the IKK inhibitor complex that, when activated, phosphorylates the inhibitor of the NF-κB transcription factors allowing for the translocation of transcription factors into the nucleus.

Generalized glucocorticoid resistance or Chrousos syndrome is a rare genetic disorder that can run in families or be sporadic. It is characterized by partial or generalized target-tissue insensitivity to glucocorticoids.
References
- ↑ Lal, Rayhan A.; Bachrach, Laura K.; Hoffman, Andrew R.; Inlora, Jingga; Rego, Shannon; Snyder, Michael P.; Lewis, David B. (2017). "A Case Report of Hypoglycemia and Hypogammaglobulinemia: DAVID Syndrome in a Patient With a Novel NFKB2 Mutation". The Journal of Clinical Endocrinology and Metabolism . 102 (7): 2127–2130. doi: 10.1210/jc.2017-00341 . PMID 28472507. S2CID 3773076.
- ↑ Brue, T.; Quentien, M. H.; Khetchoumian, K.; Bensa, M.; Capo-Chichi, J. M.; Delemer, B.; Balsalobre, A.; Nassif, C.; Papadimitriou, D. T.; Pagnier, A.; Hasselmann, C.; Patry, L.; Schwartzentruber, J.; Souchon, P. F.; Takayasu, S.; Enjalbert, A.; Van Vliet, G.; Majewski, J.; Drouin, J.; Samuels, M. E. (2014). "Mutations in NFKB2 and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies". BMC Medical Genetics . 15: 139. doi: 10.1186/s12881-014-0139-9 . PMC 4411703 . PMID 25524009.
- ↑ Lal, Rayhan A.; Bachrach, Laura K.; Hoffman, Andrew R.; Inlora, Jingga; Rego, Shannon; Snyder, Michael P.; Lewis, David B. (2017-07-01). "A Case Report of Hypoglycemia and Hypogammaglobulinemia: DAVID Syndrome in a Patient With a Novel NFKB2 Mutation". The Journal of Clinical Endocrinology & Metabolism. 102 (7): 2127–2130. doi: 10.1210/jc.2017-00341 . ISSN 0021-972X. PMID 28472507. S2CID 3773076.
- ↑ Nogueira, M.; Pinheiro, M.; Maia, R.; Silva, R. S.; Costa, C.; Campos, T.; Leão, M.; Vitor, A. B.; Castro-Correia, C.; Fontoura, M. (2020). "Symptomatic hypoglycemia in a child with common variable immunodeficiency: Deficient anterior pituitary with variable immune deficiency (DAVID) syndrome". Clinical Pediatric Endocrinology. 29 (3): 111–113. doi:10.1297/cpe.29.111. PMC 7348627 . PMID 32694887.
| | This genetic disorder article is a stub. You can help Wikipedia by expanding it. |