Related Research Articles

Bioinformatics is an interdisciplinary field that develops methods and software tools for understanding biological data, in particular when the data sets are large and complex. As an interdisciplinary field of science, bioinformatics combines biology, chemistry, physics, computer science, information engineering, mathematics and statistics to analyze and interpret the biological data. Bioinformatics has been used for in silico analyses of biological queries using computational and statistical techniques.

In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences, such as calculating the distance cost between strings in a natural language or in financial data.
The National Center for Biotechnology Information (NCBI) is part of the United States National Library of Medicine (NLM), a branch of the National Institutes of Health (NIH). It is approved and funded by the government of the United States. The NCBI is located in Bethesda, Maryland, and was founded in 1988 through legislation sponsored by US Congressman Claude Pepper.
Protein engineering is the process of developing useful or valuable proteins. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.
In bioinformatics, sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and others.
A sequence profiling tool in bioinformatics is a type of software that presents information related to a genetic sequence, gene name, or keyword input. Such tools generally take a query such as a DNA, RNA, or protein sequence or ‘keyword’ and search one or more databases for information related to that sequence. Summaries and aggregate results are provided in standardized format describing the information that would otherwise have required visits to many smaller sites or direct literature searches to compile. Many sequence profiling tools are software portals or gateways that simplify the process of finding information about a query in the large and growing number of bioinformatics databases. The access to these kinds of tools is either web based or locally downloadable executables.

Multiple sequence alignment (MSA) may refer to the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In many cases, the input set of query sequences are assumed to have an evolutionary relationship by which they share a linkage and are descended from a common ancestor. From the resulting MSA, sequence homology can be inferred and phylogenetic analysis can be conducted to assess the sequences' shared evolutionary origins. Visual depictions of the alignment as in the image at right illustrate mutation events such as point mutations that appear as differing characters in a single alignment column, and insertion or deletion mutations that appear as hyphens in one or more of the sequences in the alignment. Multiple sequence alignment is often used to assess sequence conservation of protein domains, tertiary and secondary structures, and even individual amino acids or nucleotides.
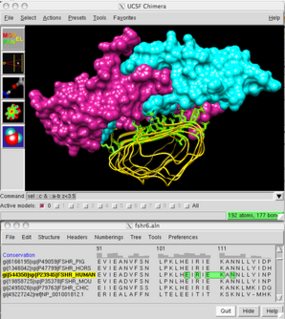
UCSF Chimera is an extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles. High-quality images and movies can be created. Chimera includes complete documentation and can be downloaded free of charge for noncommercial use.
Sequerome is a web-based sequence profiling tool for integrating the results of a BLAST sequence-alignment report with external research tools and servers that perform advanced sequence manipulations, and allowing the user to record the steps of such an analysis. Sequerome is a web-based Java tool that acts as a front-end to BLAST queries and provides simplified access to web-distributed resources for protein and nucleic acid analysis.
BLAT is a pairwise sequence alignment algorithm that was developed by Jim Kent at the University of California Santa Cruz (UCSC) in the early 2000s to assist in the assembly and annotation of the human genome. It was designed primarily to decrease the time needed to align millions of mouse genomic reads and expressed sequence tags against the human genome sequence. The alignment tools of the time were not capable of performing these operations in a manner that would allow a regular update of the human genome assembly. Compared to pre-existing tools, BLAT was ~500 times faster with performing mRNA/DNA alignments and ~50 times faster with protein/protein alignments.
The Viral Bioinformatics Resource Center (VBRC) is an online resource providing access to a database of curated viral genomes and a variety of tools for bioinformatic genome analysis. This resource was one of eight BRCs funded by NIAID with the goal of promoting research against emerging and re-emerging pathogens, particularly those seen as potential bioterrorism threats. The VBRC is now supported by Dr. Chris Upton at the University of Victoria.
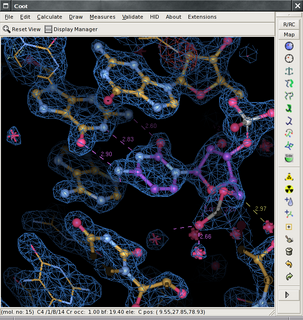
The program Coot is used to display and manipulate atomic models of macromolecules, typically of proteins or nucleic acids, using 3D computer graphics. It is primarily focused on building and validation of atomic models into three-dimensional electron density maps obtained by X-ray crystallography methods, although it has also been applied to data from electron microscopy.
UGENE is computer software for bioinformatics. It works on personal computer operating systems such as Windows, macOS, or Linux. It is released as free and open-source software, under a GNU General Public License (GPL) version 2.

HMMER is a free and commonly used software package for sequence analysis written by Sean Eddy. Its general usage is to identify homologous protein or nucleotide sequences, and to perform sequence alignments. It detects homology by comparing a profile-HMM to either a single sequence or a database of sequences. Sequences that score significantly better to the profile-HMM compared to a null model are considered to be homologous to the sequences that were used to construct the profile-HMM. Profile-HMMs are constructed from a multiple sequence alignment in the HMMER package using the hmmbuild program. The profile-HMM implementation used in the HMMER software was based on the work of Krogh and colleagues. HMMER is a console utility ported to every major operating system, including different versions of Linux, Windows, and Mac OS.
MacVector is a commercial sequence analysis application for Apple Macintosh computers running Mac OS X. It is intended to be used by molecular biologists to help analyze, design, research and document their experiments in the laboratory. MacVector 18.1 is a Universal Binary capable of running on Intel and Apple Silicon Macs.
Clone Manager is a commercial bioinformatics software work suite of Sci-Ed, that supports molecular biologists with data management and allows them to perform certain in silico preanalysis.
The Staden Package is computer software, a set of tools for DNA sequence assembly, editing, and sequence analysis. It is open-source software, released under a BSD 3-clause license.

Gene Designer is a computer software package for bioinformatics. It is used by molecular biologists from academia, government, and the pharmaceutical, chemical, agricultural, and biotechnology industries to design, clone, and validate genetic sequences. It is proprietary software, released as freeware needing registration.

In silico PCR refers to computational tools used to calculate theoretical polymerase chain reaction (PCR) results using a given set of primers (probes) to amplify DNA sequences from a sequenced genome or transcriptome.
Vector NTI was a commercial bioinformatics software package used by many life scientists in the early 2000s to work, among other things, with nucleic acids and proteins in silico. It allowed researchers to, for example, plan a DNA cloning experiment on the computer before actually performing it in the lab.
References
- ↑ "Home". bluetractorsoftware.co.uk.
- ↑ "System requirement for running DNADynamo".
- ↑ Google Scholar citations
- ↑ "About Blue Tractor Software Ltd".