
The alpha helix (α-helix) is a common motif in the secondary structure of proteins and is a right hand-helix conformation in which every backbone N−H group hydrogen bonds to the backbone C=O group of the amino acid located four residues earlier along the protein sequence.

The beta sheet, (β-sheet) is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, notably Alzheimer's disease.

A hydrogen bond is a primarily electrostatic force of attraction between a hydrogen (H) atom which is covalently bound to a more electronegative atom or group, and another electronegative atom bearing a lone pair of electrons—the hydrogen bond acceptor (Ac). Such an interacting system is generally denoted Dn–H···Ac, where the solid line denotes a polar covalent bond, and the dotted or dashed line indicates the hydrogen bond. The most frequent donor and acceptor atoms are the second-row elements nitrogen (N), oxygen (O), and fluorine (F).

In organic chemistry, a peptide bond is an amide type of covalent chemical bond linking two consecutive alpha-amino acids from C1 of one alpha-amino acid and N2 of another, along a peptide or protein chain.

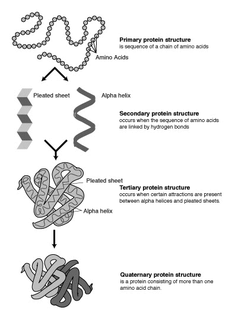
Protein secondary structure is the three dimensional form of local segments of proteins. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.

Proline (symbol Pro or P) is an organic acid classed as a proteinogenic amino acid (used in the biosynthesis of proteins), although it does not contain the amino group -NH
2 but is rather a secondary amine. The secondary amine nitrogen is in the protonated NH2+ form under biological conditions, while the carboxy group is in the deprotonated −COO− form. The "side chain" from the α carbon connects to the nitrogen forming a pyrrolidine loop, classifying it as a aliphatic amino acid. It is non-essential in humans, meaning the body can synthesize it from the non-essential amino acid L-glutamate. It is encoded by all the codons starting with CC (CCU, CCC, CCA, and CCG).
Circular dichroism (CD) is dichroism involving circularly polarized light, i.e., the differential absorption of left- and right-handed light. Left-hand circular (LHC) and right-hand circular (RHC) polarized light represent two possible spin angular momentum states for a photon, and so circular dichroism is also referred to as dichroism for spin angular momentum. This phenomenon was discovered by Jean-Baptiste Biot, Augustin Fresnel, and Aimé Cotton in the first half of the 19th century. Circular dichroism and circular birefringence are manifestations of optical activity. It is exhibited in the absorption bands of optically active chiral molecules. CD spectroscopy has a wide range of applications in many different fields. Most notably, UV CD is used to investigate the secondary structure of proteins. UV/Vis CD is used to investigate charge-transfer transitions. Near-infrared CD is used to investigate geometric and electronic structure by probing metal d→d transitions. Vibrational circular dichroism, which uses light from the infrared energy region, is used for structural studies of small organic molecules, and most recently proteins and DNA.
The hartree, also known as the Hartree energy, is the unit of energy in the Hartree atomic units system, named after the British physicist Douglas Hartree. Its CODATA recommended value is Eh = 4.3597447222071(85)×10−18 J = 27.211386245988(53) eV.
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.
The Hückel method or Hückel molecular orbital theory, proposed by Erich Hückel in 1930, is a very simple linear combination of atomic orbitals molecular orbitals method for the determination of energies of molecular orbitals of π-electrons in π-delocalized molecules, such as ethylene, benzene, butadiene, and pyridine. It is the theoretical basis for Hückel's rule for the aromaticity of π-electron cyclic, planar systems. It was later extended to conjugated molecules such as pyridine, pyrrole and furan that contain atoms other than carbon, known in this context as heteroatoms. A more dramatic extension of the method to include σ-electrons, known as the extended Hückel method (EHM), was developed by Roald Hoffmann. The extended Hückel method gives some degree of quantitative accuracy for organic molecules in general and was used to provide computational justification for the Woodward–Hoffmann rules. To distinguish the original approach from Hoffmann's extension, the Hückel method is also known as the simple Hückel method (SHM). An elementary description of the application of the simple Huckel method to benzene is given in Sections 3.4.3 and 10.2 of a student level text book.

A protein contact map represents the distance between all possible amino acid residue pairs of a three-dimensional protein structure using a binary two-dimensional matrix. For two residues and , the element of the matrix is 1 if the two residues are closer than a predetermined threshold, and 0 otherwise. Various contact definitions have been proposed: The distance between the Cα-Cα atom with threshold 6-12 Å; distance between Cβ-Cβ atoms with threshold 6-12 Å ; and distance between the side-chain centers of mass.
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.
A polyproline helix is a type of protein secondary structure which occurs in proteins comprising repeating proline residues. A left-handed polyproline II helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have trans isomers of their peptide bonds. This PPII conformation is also common in proteins and polypeptides with other amino acids apart from proline. Similarly, a more compact right-handed polyproline I helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have cis isomers of their peptide bonds. Of the twenty common naturally occurring amino acids, only proline is likely to adopt the cis isomer of the peptide bond, specifically the X-Pro peptide bond; steric and electronic factors heavily favor the trans isomer in most other peptide bonds. However, peptide bonds that replace proline with another N-substituted amino acid are also likely to adopt the cis isomer.

A pi helix is a type of secondary structure found in proteins. Discovered by crystallographer Barbara Low in 1952 and once thought to be rare, short π-helices are found in 15% of known protein structures and are believed to be an evolutionary adaptation derived by the insertion of a single amino acid into an α-helix. Because such insertions are highly destabilizing, the formation of π-helices would tend to be selected against unless it provided some functional advantage to the protein. π-helices therefore are typically found near functional sites of proteins.

A 310 helix is a type of secondary structure found in proteins and polypeptides. Of the numerous protein secondary structures present, the 310-helix is the fourth most common type observed; following α-helices, β-sheets and reverse turns. 310-helices constitute nearly 10–15% of all helices in protein secondary structures, and are typically observed as extensions of α-helices found at either their N- or C- termini. Because of the α-helices tendency to consistently fold and unfold, it has been proposed that the 310-helix serves as an intermediary conformation of sorts, and provides insight into the initiation of α-helix folding.

Davydov soliton is a quantum quasiparticle representing an excitation propagating along the protein α-helix self-trapped amide I. It is a solution of the Davydov Hamiltonian. It is named for the Soviet and Ukrainian physicist Alexander Davydov. The Davydov model describes the interaction of the amide I vibrations with the hydrogen bonds that stabilize the α-helix of proteins. The elementary excitations within the α-helix are given by the phonons which correspond to the deformational oscillations of the lattice, and the excitons which describe the internal amide I excitations of the peptide groups. Referring to the atomic structure of an α-helix region of protein the mechanism that creates the Davydov soliton can be described as follows: vibrational energy of the C=O stretching oscillators that is localized on the α-helix acts through a phonon coupling effect to distort the structure of the α-helix, while the helical distortion reacts again through phonon coupling to trap the amide I oscillation energy and prevent its dispersion. This effect is called self-localization or self-trapping. Solitons in which the energy is distributed in a fashion preserving the helical symmetry are dynamically unstable, and such symmetrical solitons once formed decay rapidly when they propagate. On the other hand, an asymmetric soliton which spontaneously breaks the local translational and helical symmetries possesses the lowest energy and is a robust localized entity.
In polymer science, the Lifson–Roig model is a helix-coil transition model applied to the alpha helix-random coil transition of polypeptides; it is a refinement of the Zimm–Bragg model that recognizes that a polypeptide alpha helix is only stabilized by a hydrogen bond only once three consecutive residues have adopted the helical conformation. To consider three consecutive residues each with two states, the Lifson–Roig model uses a 4x4 transfer matrix instead of the 2x2 transfer matrix of the Zimm–Bragg model, which considers only two consecutive residues. However, the simple nature of the coil state allows this to be reduced to a 3x3 matrix for most applications.
In protein structure, STRIDE is an algorithm for the assignment of protein secondary structure elements given the atomic coordinates of the protein, as defined by X-ray crystallography, protein NMR, or another protein structure determination method. In addition to the hydrogen bond criteria used by the more common DSSP algorithm, the STRIDE assignment criteria also include dihedral angle potentials. As such, its criteria for defining individual secondary structures are more complex than those of DSSP. The STRIDE energy function contains a hydrogen-bond term containing a Lennard-Jones-like 8-6 distance-dependent potential and two angular dependence factors reflecting the planarity of the optimized hydrogen bond geometry. The criteria for individual secondary structural elements, which are divided into the same groups as those reported by DSSP, also contain statistical probability factors derived from empirical examinations of solved structures with visually assigned secondary structure elements extracted from the Protein Data Bank.

The beta bend ribbon, or beta-bend ribbon, is a structural feature in polypeptides and proteins. The shortest possible has six amino acid residues arranged as two overlapping hydrogen bonded beta turns in which the carbonyl group of residue i is hydrogen-bonded to the NH of residue i+3 while the carbonyl group of residue i+2 is hydrogen-bonded to the NH of residue i+5. In longer ribbons, this bonding is continued in peptides of 8, 10, etc., amino acid residues. A beta bend ribbon can be regarded as an aberrant 310 helix (3/10-helix) that has lost some of its hydrogen bonds. Two websites are available to facilitate finding and examining these features in proteins: Motivated Proteins; and PDBeMotif.
Volume, Area, Dihedral Angle Reporter (VADAR) is a freely available protein structure validation web server that was developed as a collaboration between Dr. Brian Sykes and Dr. David Wishart at the University of Alberta. VADAR consists of over 15 different algorithms and programs for assessing and validating peptide and protein structures from their PDB coordinate data. VADAR is capable of determining secondary structure, identifying and classifying six different types of beta turns, determining and calculating the strength of C=O -- N-H hydrogen bonds, calculating residue-specific accessible surface areas (ASA), calculating residue volumes, determining backbone and side chain torsion angles, assessing local structure quality, evaluating global structure quality, and identifying residue "outliers". The results have been validated through extensive comparison to published data and careful visual inspection. VADAR produces both text and graphical output with most of the quantitative data presented in easily viewed tables. In particular, VADAR's output is presented in a vertical, tabular format with most of the sequence data, residue numbering and any other calculated property or feature presented from top to bottom, rather than from left to right.



