Related Research Articles
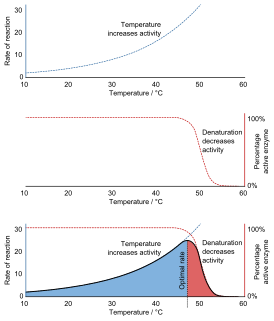
Denaturation is a process in which proteins or nucleic acids lose the quaternary structure, tertiary structure, and secondary structure which is present in their native state, by application of some external stress or compound such as a strong acid or base, a concentrated inorganic salt, an organic solvent, radiation or heat. If proteins in a living cell are denatured, this results in disruption of cell activity and possibly cell death. Protein denaturation is also a consequence of cell death. Denatured proteins can exhibit a wide range of characteristics, from conformational change and loss of solubility to aggregation due to the exposure of hydrophobic groups. Denatured proteins lose their 3D structure and therefore cannot function.

Entropy is a scientific concept, as well as a measurable physical property that is most commonly associated with a state of disorder, randomness, or uncertainty. The term and the concept are used in diverse fields, from classical thermodynamics, where it was first recognized, to the microscopic description of nature in statistical physics, and to the principles of information theory. It has found far-ranging applications in chemistry and physics, in biological systems and their relation to life, in cosmology, economics, sociology, weather science, climate change, and information systems including the transmission of information in telecommunication.

Protein folding is the physical process by which a protein chain is translated to its native three-dimensional structure, typically a "folded" conformation by which the protein becomes biologically functional. Via an expeditious and reproducible process, a polypeptide folds into its characteristic three-dimensional structure from a random coil. Each protein exists first as an unfolded polypeptide or random coil after being translated from a sequence of mRNA to a linear chain of amino acids. At this stage the polypeptide lacks any stable (long-lasting) three-dimensional structure. As the polypeptide chain is being synthesized by a ribosome, the linear chain begins to fold into its three-dimensional structure.

In chemistry, thermodynamics, and many other related fields, phase transitions are the physical processes of transition between a state of a medium, identified by some parameters, and another one, with different values of the parameters. Commonly the term is used to refer to changes among the basic states of matter: solid, liquid, and gas, as well as plasma in rare cases.

Differential scanning calorimetry (DSC) is a thermoanalytical technique in which the difference in the amount of heat required to increase the temperature of a sample and reference is measured as a function of temperature. Both the sample and reference are maintained at nearly the same temperature throughout the experiment. Generally, the temperature program for a DSC analysis is designed such that the sample holder temperature increases linearly as a function of time. The reference sample should have a well-defined heat capacity over the range of temperatures to be scanned.
The term molten globule (MG) refers to protein states that are more or less compact, but are lacking the specific tight packing of amino acid residues which creates the solid state-like tertiary structure of completely folded proteins. It was found, for example, in cytochrome c, which conserves a native-like secondary structure content but without the tightly packed protein interior, under low pH and high salt concentration. For cytochrome c and some other proteins, it has been shown that the molten globule state is a "thermodynamic state" clearly different both from the native and the denatured state, demonstrating for the first time the existence of a third equilibirum state.
In biochemistry, the native state of a protein or nucleic acid is its properly folded and/or assembled form, which is operative and functional. The native state of a biomolecule may possess all four levels of biomolecular structure, with the secondary through quaternary structure being formed from weak interactions along the covalently-bonded backbone. This is in contrast to the denatured state, in which these weak interactions are disrupted, leading to the loss of these forms of structure and retaining only the biomolecule's primary structure.
In classical statistical mechanics, the equipartition theorem relates the temperature of a system to its average energies. The equipartition theorem is also known as the law of equipartition, equipartition of energy, or simply equipartition. The original idea of equipartition was that, in thermal equilibrium, energy is shared equally among all of its various forms; for example, the average kinetic energy per degree of freedom in translational motion of a molecule should equal that in rotational motion.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, the monomers of the polymer. A single amino acid monomer may also be called a residue indicating a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo electron microscopy (cryo-EM) and dual polarisation interferometry to determine the structure of proteins.

In chemistry, a salt bridge is a combination of two non-covalent interactions: hydrogen bonding and ionic bonding. Ion pairing is one of the most important noncovalent forces in chemistry, in biological systems, in different materials and in many applications such as ion pair chromatography. It is a most commonly observed contribution to the stability to the entropically unfavorable folded conformation of proteins. Although non-covalent interactions are known to be relatively weak interactions, small stabilizing interactions can add up to make an important contribution to the overall stability of a conformer. Not only are salt bridges found in proteins, but they can also be found in supramolecular chemistry. The thermodynamics of each are explored through experimental procedures to access the free energy contribution of the salt bridge to the overall free energy of the state.

Thermostability is the quality of a substance to resist irreversible change in its chemical or physical structure, often by resisting decomposition or polymerization, at a high relative temperature.
Phi value analysis, analysis, or -value analysis is an experimental protein engineering technique for studying the structure of the folding transition state of small protein domains that fold in a two-state manner. The structure of the folding transition state is hard to find using methods like protein NMR or X-ray crystallography because folding transitions states are mobile and partly unstructured by definition. In -value analysis, the folding kinetics and conformational folding stability of the wild-type protein are compared with those of point mutants to find phi values. These measure the mutant residue's energetic contribution to the folding transition state, which reveals the degree of native structure around the mutated residue in the transition state, by accounting for the relative free energies of the unfolded state, the folded state, and the transition state for the wild-type and mutant proteins.
In biochemistry, equilibrium unfolding is the process of unfolding a protein or RNA molecule by gradually changing its environment, such as by changing the temperature or pressure, pH, adding chemical denaturants, or applying force as with an atomic force microscope tip. If the equilibrium was maintained at all steps, the process theoretically should be reversible during equilibrium folding. Equilibrium unfolding can be used to determine the thermodynamic stability of the protein or RNA structure, i.e. free energy difference between the folded and unfolded states.
A chevron plot is a way of representing protein folding kinetic data in the presence of varying concentrations of denaturant that disrupts the protein's native tertiary structure. The plot is known as "chevron" plot because of the canonical v, or chevron shape observed when the logarithm of the observed relaxation rate is plotted as a function of the denaturant concentration.

The folding funnel hypothesis is a specific version of the energy landscape theory of protein folding, which assumes that a protein's native state corresponds to its free energy minimum under the solution conditions usually encountered in cells. Although energy landscapes may be "rough", with many non-native local minima in which partially folded proteins can become trapped, the folding funnel hypothesis assumes that the native state is a deep free energy minimum with steep walls, corresponding to a single well-defined tertiary structure. The term was introduced by Ken A. Dill in a 1987 article discussing the stabilities of globular proteins.
Downhill folding is a process in which a protein folds without encountering any significant macroscopic free energy barrier. It is a key prediction of the folding funnel hypothesis of the energy landscape theory of proteins.
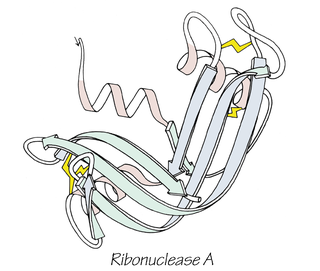
Anfinsen's dogma, also known as the thermodynamic hypothesis, is a postulate in molecular biology. It states that, at least for a small globular protein in its standard physiological environment, the native structure is determined only by the protein's amino acid sequence. The dogma was championed by the Nobel Prize Laureate Christian B. Anfinsen from his research on the folding of ribonuclease A. The postulate amounts to saying that, at the environmental conditions at which folding occurs, the native structure is a unique, stable and kinetically accessible minimum of the free energy. In other words, there are three conditions for formation of a unique protein structure:
Nucleic acid thermodynamics is the study of how temperature affects the nucleic acid structure of double-stranded DNA (dsDNA). The melting temperature (Tm) is defined as the temperature at which half of the DNA strands are in the random coil or single-stranded (ssDNA) state. Tm depends on the length of the DNA molecule and its specific nucleotide sequence. DNA, when in a state where its two strands are dissociated, is referred to as having been denatured by the high temperature.
A thermal shift assay (TSA) measures changes in the thermal denaturation temperature and hence stability of a protein under varying conditions such as variations in drug concentration, buffer pH or ionic strength, redox potential, or sequence mutation. The most common method for measuring protein thermal shifts is differential scanning fluorimetry (DSF) or thermofluor, which utilizes specialized fluorogenic dyes.
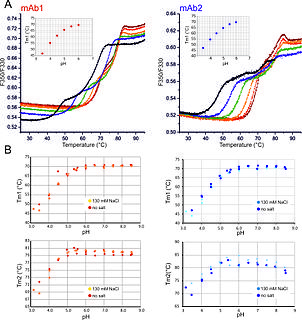
nanoDSF is a modified differential scanning fluorimetry method to determine protein stability employing intrinsic tryptophan or tyrosin fluorescence.
References
- ↑ Munoz, V.; Sanchez-Ruiz, J.M. (2004). "Exploring protein-folding ensembles: A variable-barrier model for the analysis of equilibrium unfolding experiments". Proceedings of the National Academy of Sciences. 101 (51): 17646–17651. Bibcode:2004PNAS..10117646M. doi: 10.1073/pnas.0405829101 . PMC 539728 . PMID 15591110.