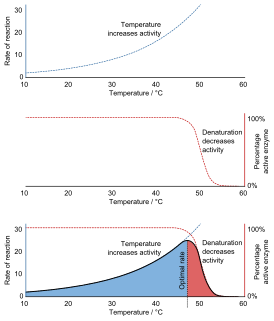
Denaturation is a process in which proteins or nucleic acids lose the quaternary structure, tertiary structure, and secondary structure which is present in their native state, by application of some external stress or compound such as a strong acid or base, a concentrated inorganic salt, an organic solvent, radiation or heat. If proteins in a living cell are denatured, this results in disruption of cell activity and possibly cell death. Protein denaturation is also a consequence of cell death. Denatured proteins can exhibit a wide range of characteristics, from conformational change and loss of solubility to aggregation due to the exposure of hydrophobic groups. Denatured proteins lose their 3D structure and therefore cannot function.

Protein folding is the physical process by which a protein chain is translated to its native three-dimensional structure, typically a "folded" conformation by which the protein becomes biologically functional. Via an expeditious and reproducible process, a polypeptide folds into its characteristic three-dimensional structure from a random coil. Each protein exists first as an unfolded polypeptide or random coil after being translated from a sequence of mRNA to a linear chain of amino acids. At this stage the polypeptide lacks any stable (long-lasting) three-dimensional structure. As the polypeptide chain is being synthesized by a ribosome, the linear chain begins to fold into its three-dimensional structure.
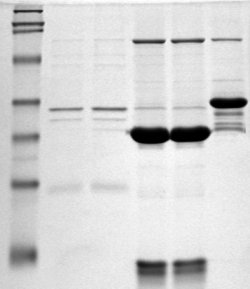
Polyacrylamide gel electrophoresis (PAGE) is a technique widely used in biochemistry, forensic chemistry, genetics, molecular biology and biotechnology to separate biological macromolecules, usually proteins or nucleic acids, according to their electrophoretic mobility. Electrophoretic mobility is a function of the length, conformation and charge of the molecule. Polyacrylamide gel electrophoresis is a powerful tool used to analyze RNA samples. When polyacrylamide gel is denatured after electrophoresis, it provides information on the sample composition of the RNA species.
The term molten globule (MG) refers to protein states that are more or less compact, but are lacking the specific tight packing of amino acid residues which creates the solid state-like tertiary structure of completely folded proteins. It was found, for example, in cytochrome c, which conserves a native-like secondary structure content but without the tightly packed protein interior, under low pH and high salt concentration. For cytochrome c and some other proteins, it has been shown that the molten globule state is a "thermodynamic state" clearly different both from the native and the denatured state, demonstrating for the first time the existence of a third equilibirum state.
Levinthal's paradox is a thought experiment, also constituting a self-reference in the theory of protein folding. In 1969, Cyrus Levinthal noted that, because of the very large number of degrees of freedom in an unfolded polypeptide chain, the molecule has an astronomical number of possible conformations. An estimate of 10300 was made in one of his papers (often incorrectly cited as the 1968 paper). For example, a polypeptide of 100 residues will have 99 peptide bonds, and therefore 198 different phi and psi bond angles. If each of these bond angles can be in one of three stable conformations, the protein may misfold into a maximum of 3198 different conformations (including any possible folding redundancy). Therefore, if a protein were to attain its correctly folded configuration by sequentially sampling all the possible conformations, it would require a time longer than the age of the universe to arrive at its correct native conformation. This is true even if conformations are sampled at rapid (nanosecond or picosecond) rates. The "paradox" is that most small proteins fold spontaneously on a millisecond or even microsecond time scale. The solution to this paradox has been established by computational approaches to protein structure prediction.

Enzyme assays are laboratory methods for measuring enzymatic activity. They are vital for the study of enzyme kinetics and enzyme inhibition.

In chemistry, a salt bridge is a combination of two non-covalent interactions: hydrogen bonding and ionic bonding. Ion pairing is one of the most important noncovalent forces in chemistry, in biological systems, in different materials and in many applications such as ion pair chromatography. It is a most commonly observed contribution to the stability to the entropically unfavorable folded conformation of proteins. Although non-covalent interactions are known to be relatively weak interactions, small stabilizing interactions can add up to make an important contribution to the overall stability of a conformer. Not only are salt bridges found in proteins, but they can also be found in supramolecular chemistry. The thermodynamics of each are explored through experimental procedures to access the free energy contribution of the salt bridge to the overall free energy of the state.

Enzyme kinetics is the study of the rates of enzyme-catalysed chemical reactions. In enzyme kinetics, the reaction rate is measured and the effects of varying the conditions of the reaction are investigated. Studying an enzyme's kinetics in this way can reveal the catalytic mechanism of this enzyme, its role in metabolism, how its activity is controlled, and how a drug or a modifier might affect the rate.
The temperature jump method is a technique used in chemical kinetics for the measurement of very rapid reaction rates. It is one of a class of chemical relaxation methods pioneered by the German physical chemist Manfred Eigen in the 1950s. In these methods, a reacting system initially at equilibrium is perturbed rapidly and then observed as it relaxes back to equilibrium. In the case of temperature jump, the perturbation involves rapid heating which changes the value of the equilibrium constant, followed by relaxation to equilibrium at the new temperature.
Phi value analysis, analysis, or -value analysis is an experimental protein engineering technique for studying the structure of the folding transition state of small protein domains that fold in a two-state manner. The structure of the folding transition state is hard to find using methods like protein NMR or X-ray crystallography because folding transitions states are mobile and partly unstructured by definition. In -value analysis, the folding kinetics and conformational folding stability of the wild-type protein are compared with those of point mutants to find phi values. These measure the mutant residue's energetic contribution to the folding transition state, which reveals the degree of native structure around the mutated residue in the transition state, by accounting for the relative free energies of the unfolded state, the folded state, and the transition state for the wild-type and mutant proteins.
In biochemistry, equilibrium unfolding is the process of unfolding a protein or RNA molecule by gradually changing its environment, such as by changing the temperature or pressure, pH, adding chemical denaturants, or applying force as with an atomic force microscope tip. If the equilibrium was maintained at all steps, the process theoretically should be reversible during equilibrium folding. Equilibrium unfolding can be used to determine the thermodynamic stability of the protein or RNA structure, i.e. free energy difference between the folded and unfolded states.
Denaturation midpoint of a protein is defined as the temperature (Tm) or concentration of denaturant (Cm) at which both the folded and unfolded states are equally populated at equilibrium. Tm is often determined using a thermal shift assay.
Downhill folding is a process in which a protein folds without encountering any significant macroscopic free energy barrier. It is a key prediction of the folding funnel hypothesis of the energy landscape theory of proteins.
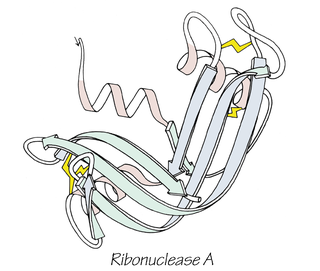
Anfinsen's dogma, also known as the thermodynamic hypothesis, is a postulate in molecular biology. It states that, at least for a small globular protein in its standard physiological environment, the native structure is determined only by the protein's amino acid sequence. The dogma was championed by the Nobel Prize Laureate Christian B. Anfinsen from his research on the folding of ribonuclease A. The postulate amounts to saying that, at the environmental conditions at which folding occurs, the native structure is a unique, stable and kinetically accessible minimum of the free energy. In other words, there are three conditions for formation of a unique protein structure:
Single molecule fluorescence resonance energy transfer is a biophysical technique used to measure distances at the 1-10 nanometer scale in single molecules, typically biomolecules. It is an application of FRET wherein a pair of donor and acceptor fluorophores are excited and detected on a single molecule level. In contrast to "ensemble FRET" which provides the FRET signal of a high number of molecules, single-molecule FRET is able to resolve the FRET signal of each individual molecule. The variation of the smFRET signal is useful to reveal kinetic information that an ensemble measurement cannot provide, especially when the system is under equilibrium. Heterogeneity among different molecules can also be observed.
Pressure jump is a technique used in the study of chemical kinetics. It involves making rapid changes to the pressure of an experimental system and observing the return to equilibrium or steady state. This allows the study of the shift in equilibrium of reactions that equilibrate in periods between milliseconds to hours, these changes often being observed using absorption spectroscopy, or fluorescence spectroscopy though other spectroscopic techniques such as CD, FTIR or NMR can also be used.
In chemistry, reaction progress kinetic analysis (RPKA) is a subset of a broad range of kinetic techniques utilized to determine the rate laws of chemical reactions and to aid in elucidation of reaction mechanisms. While the concepts guiding reaction progress kinetic analysis are not new, the process was formalized by Professor Donna Blackmond in the late 1990s and has since seen increasingly widespread use. Unlike more common pseudo-first-order analysis, in which an overwhelming excess of one or more reagents is used relative to a species of interest, RPKA probes reactions at synthetically relevant conditions Generally, this analysis involves a system in which the concentrations of multiple reactants are changing measurably over the course of the reaction. As the mechanism can vary depending on the relative and absolute concentrations of the species involved, this approach obtains results that are much more representative of reaction behavior under commonly utilized conditions than do traditional tactics. Furthermore, information obtained by observation of the reaction over time may provide insight regarding unexpected behavior such as induction periods, catalyst deactivation, or changes in mechanism.
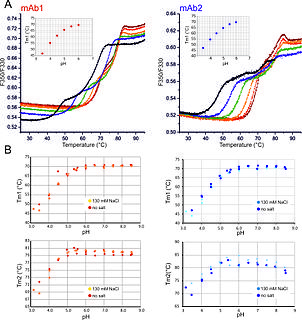
nanoDSF is a modified differential scanning fluorimetry method to determine protein stability employing intrinsic tryptophan or tyrosin fluorescence.
Jayant Bhalchandra Udgaonkar is an Indian biochemist, molecular biologist, academic and the Director of the Indian Institute of Science Education and Research, Pune. He was previously a senior professor at the National Centre for Biological Sciences. A J. C. Bose National Fellow, he is known for his studies on protein folding. He is an elected fellow of the Indian Academy of Sciences, Indian National Science Academy and The World Academy of Sciences. The Council of Scientific and Industrial Research, the apex agency of the Government of India for scientific research, awarded him the Shanti Swarup Bhatnagar Prize for Science and Technology, one of the highest Indian science awards, in 2000, for his contributions to biological sciences. He is the son of noted scientist Padmabhushan Bhalchandra Udgaonkar.
George Paul Hess was a research biochemist who specialized in studying acetylcholine receptors. Hess developed laser pulse photolysis and a quench flow technique.