
The polymerase chain reaction (PCR) is a method widely used to make millions to billions of copies of a specific DNA sample rapidly, allowing scientists to amplify a very small sample of DNA sufficiently to enable detailed study. PCR was invented in 1983 by American biochemist Kary Mullis at Cetus Corporation. Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.

A DNA sequencer is a scientific instrument used to automate the DNA sequencing process. Given a sample of DNA, a DNA sequencer is used to determine the order of the four bases: G (guanine), C (cytosine), A (adenine) and T (thymine). This is then reported as a text string, called a read. Some DNA sequencers can be also considered optical instruments as they analyze light signals originating from fluorochromes attached to nucleotides.

DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.

Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. It was first commercialized by Applied Biosystems in 1986. More recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies in that it can produce DNA sequence reads of > 500 nucleotides and maintains a very low error rate with accuracies around 99.99%. Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2 as well as for the surveillance of norovirus outbreaks through the Center for Disease Control and Prevention's (CDC) CaliciNet surveillance network.
The overlap extension polymerase chain reaction is a variant of PCR. It is also referred to as Splicing by overlap extension / Splicing by overhang extension (SOE) PCR. It is used assemble multiple smaller double stranded DNA fragments into a larger DNA sequence. OE-PCR is widely used to insert mutations at specific points in a sequence or to assemble custom DNA sequence from smaller DNA fragments into a larger polynucleotide.
SNP genotyping is the measurement of genetic variations of single nucleotide polymorphisms (SNPs) between members of a species. It is a form of genotyping, which is the measurement of more general genetic variation. SNPs are one of the most common types of genetic variation. An SNP is a single base pair mutation at a specific locus, usually consisting of two alleles. SNPs are found to be involved in the etiology of many human diseases and are becoming of particular interest in pharmacogenetics. Because SNPs are conserved during evolution, they have been proposed as markers for use in quantitative trait loci (QTL) analysis and in association studies in place of microsatellites. The use of SNPs is being extended in the HapMap project, which aims to provide the minimal set of SNPs needed to genotype the human genome. SNPs can also provide a genetic fingerprint for use in identity testing. The increase of interest in SNPs has been reflected by the furious development of a diverse range of SNP genotyping methods.
An allele-specific oligonucleotide (ASO) is a short piece of synthetic DNA complementary to the sequence of a variable target DNA. It acts as a probe for the presence of the target in a Southern blot assay or, more commonly, in the simpler dot blot assay. It is a common tool used in genetic testing, forensics, and molecular biology research.

2 Base Encoding, also called SOLiD, is a next-generation sequencing technology developed by Applied Biosystems and has been commercially available since 2008. These technologies generate hundreds of thousands of small sequence reads at one time. Well-known examples of such DNA sequencing methods include 454 pyrosequencing, the Solexa system and the SOLiD system. These methods have reduced the cost from $0.01/base in 2004 to nearly $0.0001/base in 2006 and increased the sequencing capacity from 1,000,000 bases/machine/day in 2004 to more than 100,000,000 bases/machine/day in 2006.
Molecular Inversion Probe (MIP) belongs to the class of Capture by Circularization molecular techniques for performing genomic partitioning, a process through which one captures and enriches specific regions of the genome. Probes used in this technique are single stranded DNA molecules and, similar to other genomic partitioning techniques, contain sequences that are complementary to the target in the genome; these probes hybridize to and capture the genomic target. MIP stands unique from other genomic partitioning strategies in that MIP probes share the common design of two genomic target complementary segments separated by a linker region. With this design, when the probe hybridizes to the target, it undergoes an inversion in configuration and circularizes. Specifically, the two target complementary regions at the 5’ and 3’ ends of the probe become adjacent to one another while the internal linker region forms a free hanging loop. The technology has been used extensively in the HapMap project for large-scale SNP genotyping as well as for studying gene copy alterations and characteristics of specific genomic loci to identify biomarkers for different diseases such as cancer. Key strengths of the MIP technology include its high specificity to the target and its scalability for high-throughput, multiplexed analyses where tens of thousands of genomic loci are assayed simultaneously.
COLD-PCR is a modified polymerase chain reaction (PCR) protocol that enriches variant alleles from a mixture of wildtype and mutation-containing DNA. The ability to preferentially amplify and identify minority alleles and low-level somatic DNA mutations in the presence of excess wildtype alleles is useful for the detection of mutations. Detection of mutations is important in the case of early cancer detection from tissue biopsies and body fluids such as blood plasma or serum, assessment of residual disease after surgery or chemotherapy, disease staging and molecular profiling for prognosis or tailoring therapy to individual patients, and monitoring of therapy outcome and cancer remission or relapse. Common PCR will amplify both the major (wildtype) and minor (mutant) alleles with the same efficiency, occluding the ability to easily detect the presence of low-level mutations. The capacity to detect a mutation in a mixture of variant/wildtype DNA is valuable because this mixture of variant DNAs can occur when provided with a heterogeneous sample – as is often the case with cancer biopsies. Currently, traditional PCR is used in tandem with a number of different downstream assays for genotyping or the detection of somatic mutations. These can include the use of amplified DNA for RFLP analysis, MALDI-TOF genotyping, or direct sequencing for detection of mutations by Sanger sequencing or pyrosequencing. Replacing traditional PCR with COLD-PCR for these downstream assays will increase the reliability in detecting mutations from mixed samples, including tumors and body fluids.
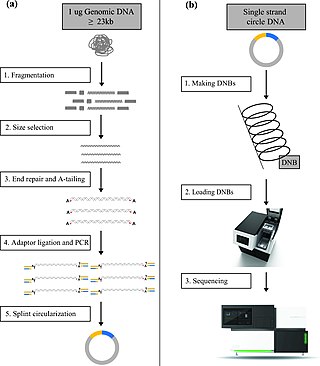
DNA nanoball sequencing is a high throughput sequencing technology that is used to determine the entire genomic sequence of an organism. The method uses rolling circle replication to amplify small fragments of genomic DNA into DNA nanoballs. Fluorescent nucleotides bind to complementary nucleotides and are then polymerized to anchor sequences bound to known sequences on the DNA template. The base order is determined via the fluorescence of the bound nucleotides This DNA sequencing method allows large numbers of DNA nanoballs to be sequenced per run at lower reagent costs compared to other next generation sequencing platforms. However, a limitation of this method is that it generates only short sequences of DNA, which presents challenges to mapping its reads to a reference genome. After purchasing Complete Genomics, the Beijing Genomics Institute (BGI) refined DNA nanoball sequencing to sequence nucleotide samples on their own platform.
Massive parallel sequencing or massively parallel sequencing is any of several high-throughput approaches to DNA sequencing using the concept of massively parallel processing; it is also called next-generation sequencing (NGS) or second-generation sequencing. Some of these technologies emerged between 1993 and 1998 and have been commercially available since 2005. These technologies use miniaturized and parallelized platforms for sequencing of 1 million to 43 billion short reads per instrument run.

Illumina dye sequencing is a technique used to determine the series of base pairs in DNA, also known as DNA sequencing. The reversible terminated chemistry concept was invented by Bruno Canard and Simon Sarfati at the Pasteur Institute in Paris. It was developed by Shankar Balasubramanian and David Klenerman of Cambridge University, who subsequently founded Solexa, a company later acquired by Illumina. This sequencing method is based on reversible dye-terminators that enable the identification of single nucleotides as they are washed over DNA strands. It can also be used for whole-genome and region sequencing, transcriptome analysis, metagenomics, small RNA discovery, methylation profiling, and genome-wide protein-nucleic acid interaction analysis.
Multiple Annealing and Looping Based Amplification Cycles (MALBAC) is a quasilinear whole genome amplification method. Unlike conventional DNA amplification methods that are non-linear or exponential, MALBAC utilizes special primers that allow amplicons to have complementary ends and therefore to loop, preventing DNA from being copied exponentially. This results in amplification of only the original genomic DNA and therefore reduces amplification bias. MALBAC is “used to create overlapped shotgun amplicons covering most of the genome”. For next generation sequencing, MALBAC is followed by regular PCR which is used to further amplify amplicons.
Magnetic sequencing is a single-molecule sequencing method in development. A DNA hairpin, containing the sequence of interest, is bound between a magnetic bead and a glass surface. A magnetic field is applied to stretch the hairpin open into single strands, and the hairpin refolds after decreasing of the magnetic field. The hairpin length can be determined by direct imaging of the diffraction rings of the magnetic beads using a simple microscope. The DNA sequences are determined by measuring the changes in the hairpin length following successful hybridization of complementary nucleotides.
SNV calling from NGS data is any of a range of methods for identifying the existence of single nucleotide variants (SNVs) from the results of next generation sequencing (NGS) experiments. These are computational techniques, and are in contrast to special experimental methods based on known population-wide single nucleotide polymorphisms. Due to the increasing abundance of NGS data, these techniques are becoming increasingly popular for performing SNP genotyping, with a wide variety of algorithms designed for specific experimental designs and applications. In addition to the usual application domain of SNP genotyping, these techniques have been successfully adapted to identify rare SNPs within a population, as well as detecting somatic SNVs within an individual using multiple tissue samples.

Whole genome bisulfite sequencing is a next-generation sequencing technology used to determine the DNA methylation status of single cytosines by treating the DNA with sodium bisulfite before high-throughput DNA sequencing. The DNA methylation status at various genes can reveal information regarding gene regulation and transcriptional activities. This technique was developed in 2009 along with reduced representation bisulfite sequencing after bisulfite sequencing became the gold standard for DNA methylation analysis.

In epitranscriptomic sequencing, most methods focus on either (1) enrichment and purification of the modified RNA molecules before running on the RNA sequencer, or (2) improving or modifying bioinformatics analysis pipelines to call the modification peaks. Most methods have been adapted and optimized for mRNA molecules, except for modified bisulfite sequencing for profiling 5-methylcytidine which was optimized for tRNAs and rRNAs.
BLESS, also known as breaks labeling, enrichment on streptavidin and next-generation sequencing, is a method used to detect genome-wide double-strand DNA damage. In contrast to chromatin immunoprecipitation (ChIP)-based methods of identifying DNA double-strand breaks (DSBs) by labeling DNA repair proteins, BLESS utilizes biotinylated DNA linkers to directly label genomic DNA in situ which allows for high-specificity enrichment of samples on streptavidin beads and the subsequent sequencing-based DSB mapping to nucleotide resolution.
GUIDE-Seq is a molecular biology technique that allows for the unbiased in vitro detection of off-target genome editing events in DNA caused by CRISPR/Cas9 as well as other RNA-guided nucleases in living cells. Similar to LAM-PCR, it employs multiple PCRs to amplify regions of interest that contain a specific insert that preferentially integrates into double-stranded breaks. As gene therapy is an emerging field, GUIDE-Seq has gained traction as a cheap method to detect the off-target effects of potential therapeutics without needing whole genome sequencing.

