
A prion is a misfolded protein that can transmit its misfoldedness to normal variants of the same protein and trigger cellular death. Prions cause prion diseases known as transmissible spongiform encephalopathies (TSEs) that are transmissible, fatal neurodegenerative diseases in humans and animals. The proteins may misfold sporadically, due to genetic mutations, or by exposure to an already misfolded protein. The consequent abnormal three-dimensional structure confers on them the ability to cause misfolding of other proteins.

Protein folding is the physical process where a protein chain is translated into its native three-dimensional structure, typically a "folded" conformation, by which the protein becomes biologically functional. Via an expeditious and reproducible process, a polypeptide folds into its characteristic three-dimensional structure from a random coil. Each protein exists first as an unfolded polypeptide or random coil after being translated from a sequence of mRNA into a linear chain of amino acids. At this stage, the polypeptide lacks any stable three-dimensional structure. As the polypeptide chain is being synthesized by a ribosome, the linear chain begins to fold into its three-dimensional structure.

In molecular biology, molecular chaperones are proteins that assist the conformational folding or unfolding of large proteins or macromolecular protein complexes. There are a number of classes of molecular chaperones, all of which function to assist large proteins in proper protein folding during or after synthesis, and after partial denaturation. Chaperones are also involved in the translocation of proteins for proteolysis.

Amyloids are aggregates of proteins characterised by a fibrillar morphology of typically 7–13 nm in diameter, a β-sheet secondary structure and ability to be stained by particular dyes, such as Congo red. In the human body, amyloids have been linked to the development of various diseases. Pathogenic amyloids form when previously healthy proteins lose their normal structure and physiological functions (misfolding) and form fibrous deposits within and around cells. These protein misfolding and deposition processes disrupt the healthy function of tissues and organs.

Alpha-synuclein (aSyn) is a protein that, in humans, is encoded by the SNCA gene. Alpha-synuclein is a neuronal protein that regulates synaptic vesicle trafficking and subsequent neurotransmitter release.
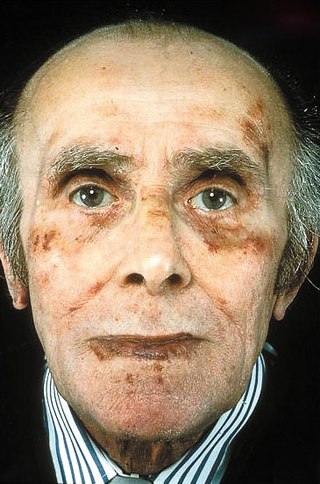
Amyloidosis is a group of diseases in which abnormal proteins, known as amyloid fibrils, build up in tissue. There are several non-specific and vague signs and symptoms associated with amyloidosis. These include fatigue, peripheral edema, weight loss, shortness of breath, palpitations, and feeling faint with standing. In AL amyloidosis, specific indicators can include enlargement of the tongue and periorbital purpura. In wild-type ATTR amyloidosis, non-cardiac symptoms include: bilateral carpal tunnel syndrome, lumbar spinal stenosis, biceps tendon rupture, small fiber neuropathy, and autonomic dysfunction.

Transthyretin (TTR or TBPA) is a transport protein in the plasma and cerebrospinal fluid that transports the thyroid hormone thyroxine (T4) and retinol to the liver. This is how transthyretin gained its name: transports thyroxine and retinol. The liver secretes TTR into the blood, and the choroid plexus secretes TTR into the cerebrospinal fluid.
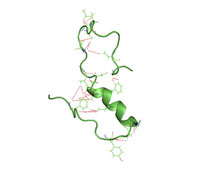
Amyloid beta denotes peptides of 36–43 amino acids that are the main component of the amyloid plaques found in the brains of people with Alzheimer's disease. The peptides derive from the amyloid-beta precursor protein (APP), which is cleaved by beta secretase and gamma secretase to yield Aβ in a cholesterol-dependent process and substrate presentation. Aβ molecules can aggregate to form flexible soluble oligomers which may exist in several forms. It is now believed that certain misfolded oligomers can induce other Aβ molecules to also take the misfolded oligomeric form, leading to a chain reaction akin to a prion infection. The oligomers are toxic to nerve cells. The other protein implicated in Alzheimer's disease, tau protein, also forms such prion-like misfolded oligomers, and there is some evidence that misfolded Aβ can induce tau to misfold.

Cardiac amyloidosis is a subcategory of amyloidosis where there is depositing of the protein amyloid in the cardiac muscle and surrounding tissues. Amyloid, a misfolded and insoluble protein, can become a deposit in the heart's atria, valves, or ventricles. These deposits can cause thickening of different sections of the heart, leading to decreased cardiac function. The overall decrease in cardiac function leads to a plethora of symptoms. This multisystem disease was often misdiagnosed, with diagnosis previously occurring after death during autopsy. However, recent advancements of technologies have increased the diagnosis of the disease. Cardiac amyloidosis has multiple sub-types including light chain, familial, and senile. One of the most studied types is light chain cardiac amyloidosis. Prognosis depends on the extent of the deposits in the body and the type of amyloidosis. New treatment methods are actively being researched in regards to the treatment of heart failure and specific cardiac amyloidosis problems.

Alpha sheet is an atypical secondary structure in proteins, first proposed by Linus Pauling and Robert Corey in 1951. The hydrogen bonding pattern in an alpha sheet is similar to that of a beta sheet, but the orientation of the carbonyl and amino groups in the peptide bond units is distinctive; in a single strand, all the carbonyl groups are oriented in the same direction on one side of the pleat, and all the amino groups are oriented in the same direction on the opposite side of the sheet. Thus the alpha sheet accumulates an inherent separation of electrostatic charge, with one edge of the sheet exposing negatively charged carbonyl groups and the opposite edge exposing positively charged amino groups. Unlike the alpha helix and beta sheet, the alpha sheet configuration does not require all component amino acid residues to lie within a single region of dihedral angles; instead, the alpha sheet contains residues of alternating dihedrals in the traditional right-handed (αR) and left-handed (αL) helical regions of Ramachandran space. Although the alpha sheet is only rarely observed in natural protein structures, it has been speculated to play a role in amyloid disease and it was found to be a stable form for amyloidogenic proteins in molecular dynamics simulations. Alpha sheets have also been observed in X-ray crystallography structures of designed peptides.

In medicine, proteinopathy, or proteopathy, protein conformational disorder, or protein misfolding disease, is a class of diseases in which certain proteins become structurally abnormal, and thereby disrupt the function of cells, tissues and organs of the body. Often the proteins fail to fold into their normal configuration; in this misfolded state, the proteins can become toxic in some way or they can lose their normal function. The proteinopathies include such diseases as Creutzfeldt–Jakob disease and other prion diseases, Alzheimer's disease, Parkinson's disease, amyloidosis, multiple system atrophy, and a wide range of other disorders. The term proteopathy was first proposed in 2000 by Lary Walker and Harry LeVine.

Acylphosphatase-2 is an enzyme that in humans is encoded by the ACYP2 gene.
The familial amyloid neuropathies are a rare group of autosomal dominant diseases wherein the autonomic nervous system and/or other nerves are compromised by protein aggregation and/or amyloid fibril formation.

In molecular biology, protein aggregation is a phenomenon in which intrinsically-disordered or mis-folded proteins aggregate either intra- or extracellularly. Protein aggregates have been implicated in a wide variety of diseases known as amyloidoses, including ALS, Alzheimer's, Parkinson's and prion disease.

Sir Christopher Martin Dobson was a British chemist, who was the John Humphrey Plummer Professor of Chemical and Structural Biology in the Department of Chemistry at the University of Cambridge, and Master of St John's College, Cambridge.
Edward Claus Franklin was a pioneering American immunologist and physician. He made major gains in the study of the aging process with contributions that led to the discovery of a group of abnormal protein aggregates known as amyloids, and played a key role in the fight against Arthritis, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, Diabetes, and Cardiac arrhythmia. Franklin was a professor of medicine at the New York University School of Medicine, a member of the National Academy of Sciences, president of the American Society for Clinical Investigation, and director of Irvington House Institute. The New York Times called Franklin "an international authority on the human immune system".
Sheena Elizabeth Radford FRS FMedSci is a British biophysicist, and Astbury Professor of Biophysics in the Astbury Centre for Structural Molecular Biology, School of Molecular and Cellular Biology at the University of Leeds. Radford is the Associate Editor of the Journal of Molecular Biology.
Jayant Bhalchandra Udgaonkar is an Indian biochemist, molecular biologist, academic and the director of the Indian Institute of Science Education and Research, Pune. He was previously a senior professor at the National Centre for Biological Sciences. A J. C. Bose National Fellow, he is known for his studies on protein folding. He is an elected fellow of the Indian Academy of Sciences, Indian National Science Academy and The World Academy of Sciences. The Council of Scientific and Industrial Research, the apex agency of the Government of India for scientific research, awarded him the Shanti Swarup Bhatnagar Prize for Science and Technology, one of the highest Indian science awards, in 2000, for his contributions to biological sciences. He is the son of noted scientist Padmabhushan Bhalchandra Udgaonkar.

Michele Vendruscolo is an Italian British physicist working in the UK, noted for his theoretical and experimental work on protein folding, misfolding and aggregation.
Jean Baum is an American chemist. She is the distinguished professor of chemistry and chemical biology at Rutgers University, where she is also vice dean for research and graduate education in the school of arts and sciences, and also vice chair of the department of chemistry and chemical biology. Her research investigates protein–protein interaction and protein aggregation using nuclear magnetic resonance spectroscopy (NMR) and other biochemical and biophysical techniques. She serves as treasurer for the Protein Society.
