
Streptococcal pharyngitis, also known as streptococcal sore throat, is pharyngitis caused by Streptococcus pyogenes, a gram-positive, group A streptococcus. Common symptoms include fever, sore throat, red tonsils, and enlarged lymph nodes in the front of the neck. A headache and nausea or vomiting may also occur. Some develop a sandpaper-like rash which is known as scarlet fever. Symptoms typically begin one to three days after exposure and last seven to ten days.

A cough is a sudden expulsion of air through the large breathing passages which can help clear them of fluids, irritants, foreign particles and microbes. As a protective reflex, coughing can be repetitive with the cough reflex following three phases: an inhalation, a forced exhalation against a closed glottis, and a violent release of air from the lungs following opening of the glottis, usually accompanied by a distinctive sound.

Bronchiectasis is a disease in which there is permanent enlargement of parts of the airways of the lung. Symptoms typically include a chronic cough with mucus production. Other symptoms include shortness of breath, coughing up blood, and chest pain. Wheezing and nail clubbing may also occur. Those with the disease often get lung infections.

An upper respiratory tract infection (URTI) is an illness caused by an acute infection, which involves the upper respiratory tract, including the nose, sinuses, pharynx, larynx or trachea. This commonly includes nasal obstruction, sore throat, tonsillitis, pharyngitis, laryngitis, sinusitis, otitis media, and the common cold. Most infections are viral in nature, and in other instances, the cause is bacterial. URTIs can also be fungal or helminthic in origin, but these are less common.
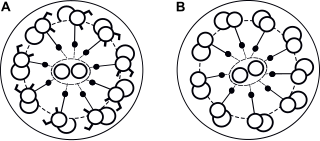
Primary ciliary dyskinesia (PCD) is a rare, autosomal recessive genetic ciliopathy, that causes defects in the action of cilia lining the upper and lower respiratory tract, sinuses, Eustachian tube, middle ear, fallopian tube, and flagella of sperm cells. The alternative name of "immotile ciliary syndrome" is no longer favored as the cilia do have movement, but are merely inefficient or unsynchronized. When accompanied by situs inversus the condition is known as Kartagener syndrome.

Granulomatosis with polyangiitis (GPA), previously known as Wegener's granulomatosis (WG), after the German physician Friedrich Wegener, is a rare long-term systemic disorder that involves the formation of granulomas and inflammation of blood vessels (vasculitis). It is an autoimmune disease and a form of vasculitis that affects small- and medium-size vessels in many organs but most commonly affects the upper respiratory tract, lungs and kidneys. The signs and symptoms of GPA are highly varied and reflect which organs are supplied by the affected blood vessels. Typical signs and symptoms include nosebleeds, stuffy nose and crustiness of nasal secretions, and inflammation of the uveal layer of the eye. Damage to the heart, lungs and kidneys can be fatal.

Lower respiratory tract infection (LRTI) is a term often used as a synonym for pneumonia but can also be applied to other types of infection including lung abscess and acute bronchitis. Symptoms include shortness of breath, weakness, fever, coughing and fatigue. A routine chest X-ray is not always necessary for people who have symptoms of a lower respiratory tract infection.

Aspirin-exacerbated respiratory disease (AERD), also called NSAID-exacerbated respiratory disease (N-ERD) or historically aspirin-induced asthma and Samter's Triad, is a long-term disease defined by three simultaneous symptoms: asthma, chronic rhinosinusitis with nasal polyps, and intolerance of aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs). Compared to aspirin tolerant patients, AERD patients' asthma and nasal polyps are generally more severe. Reduction or loss of the ability to smell is extremely common, occurring in more than 90% of people with the disease. AERD most commonly begins in early- to mid-adulthood and has no known cure. While NSAID intolerance is a defining feature of AERD, avoidance of NSAIDs does not affect the onset, development or perennial nature of the disease.

Respiratory diseases, or lung diseases, are pathological conditions affecting the organs and tissues that make gas exchange difficult in air-breathing animals. They include conditions of the respiratory tract including the trachea, bronchi, bronchioles, alveoli, pleurae, pleural cavity, the nerves and muscles of respiration. Respiratory diseases range from mild and self-limiting, such as the common cold, influenza, and pharyngitis to life-threatening diseases such as bacterial pneumonia, pulmonary embolism, tuberculosis, acute asthma, lung cancer, and severe acute respiratory syndromes, such as COVID-19. Respiratory diseases can be classified in many different ways, including by the organ or tissue involved, by the type and pattern of associated signs and symptoms, or by the cause of the disease.
Bronchoalveolar lavage (BAL), also known as bronchoalveolar washing, is a diagnostic method of the lower respiratory system in which a bronchoscope is passed through the mouth or nose into an appropriate airway in the lungs, with a measured amount of fluid introduced and then collected for examination. This method is typically performed to diagnose pathogenic infections of the lower respiratory airways, though it also has been shown to have utility in diagnosing interstitial lung disease. Bronchoalveolar lavage can be a more sensitive method of detection than nasal swabs in respiratory molecular diagnostics, as has been the case with SARS-CoV-2 where bronchoalveolar lavage samples detect copies of viral RNA after negative nasal swab testing.

Barakat syndrome is a rare disease characterized by hypoparathyroidism, sensorineural deafness and renal disease, and hence also known as HDR syndrome. It is an autosomal dominant condition with incomplete penetrance and variable expressivity that was first described by Amin J. Barakat et al. in 1977.

Intestinal pseudo-obstruction (IPO) is a clinical syndrome caused by severe impairment in the ability of the intestines to push food through. It is characterized by the signs and symptoms of intestinal obstruction without any lesion in the intestinal lumen. Clinical features mimic those seen with mechanical intestinal obstructions and can include abdominal pain, nausea, abdominal distension, vomiting, dysphagia and constipation depending upon the part of the gastrointestinal tract involved.
Idiopathic pulmonary haemosiderosis (IPH) is a lung disease of unknown cause that is characterized by alveolar capillary bleeding and accumulation of haemosiderin in the lungs. It is rare, with an incidence between 0.24 and 1.23 cases per million people.
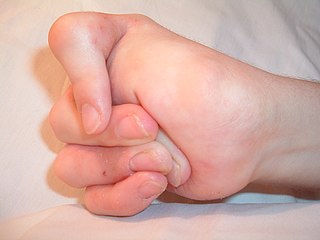
Freeman–Sheldon syndrome (FSS) is a very rare form of multiple congenital contracture (MCC) syndromes (arthrogryposes) and is the most severe form of distal arthrogryposis (DA). It was originally described by Ernest Arthur Freeman and Joseph Harold Sheldon in 1938.
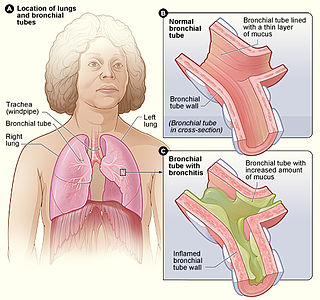
Bronchitis is inflammation of the bronchi in the lungs that causes coughing. Bronchitis usually begins as an infection in the nose, ears, throat, or sinuses. The infection then makes its way down to the bronchi. Symptoms include coughing up sputum, wheezing, shortness of breath, and chest pain. Bronchitis can be acute or chronic.
Williams–Campbell syndrome (WCS) is a disease of the airways where cartilage in the bronchi is defective. It is a form of congenital cystic bronchiectasis. This leads to collapse of the airways and bronchiectasis. It acts as one of the differential to allergic bronchopulmonary aspergillosis. WCS is a deficiency of the bronchial cartilage distally.

Stimmler syndrome is a rare autosomal recessive congenital disorder first described by Stimmler et al. in 1970. It is characterized by dwarfism, diabetes, a small head, and high levels of alanine in the urine.
Cerebro-costo-mandibular syndrome is a very rare genetic disorder which is characterized by jaw/chin, palate and rib abnormalities.
Familial benign copper deficiency, also known as Familial benign hypocupremia is a rare genetic disorder which is characterized by hypocupremia that causes symptoms such as epilepsy, hypotonia, seborrheic skin, thriving failure and mild anemia. Radiological findings include tibia and femur spurring. Transmission is thought to be either autosomal dominant or X-linked dominant.
Ankylosing vertebral hyperostosis with tylosis is a rare autosomal dominant genetic disorder characterized by ossification of the paraspinal ligament, sclerosis of the sacroiliac joint, and punctate hyperkeratosis. Some people with the condition are actually asymptomatic, which means they're relatively unaffected by it, the people who do show symptoms of it usually only show chronic/recurring back pain ranging from mild to moderate and, occasionally, obesity. It has only been described in 8 members of a 2-generation Greek Cypriot family. It is a type of dysostosis.
