Related Research Articles

Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

Alternative splicing, or alternative RNA splicing, or differential splicing, is an alternative splicing process during gene expression that allows a single gene to code for multiple proteins. In this process, particular exons of a gene may be included within or excluded from the final, processed messenger RNA (mRNA) produced from that gene. This means the exons are joined in different combinations, leading to different (alternative) mRNA strands. Consequently, the proteins translated from alternatively spliced mRNAs usually contain differences in their amino acid sequence and, often, in their biological functions.
In computational biology, gene prediction or gene finding refers to the process of identifying the regions of genomic DNA that encode genes. This includes protein-coding genes as well as RNA genes, but may also include prediction of other functional elements such as regulatory regions. Gene finding is one of the first and most important steps in understanding the genome of a species once it has been sequenced.
In molecular biology, open reading frames (ORFs) are defined as spans of DNA sequence between the start and stop codons. Usually, this is considered within a studied region of a prokaryotic DNA sequence, where only one of the six possible reading frames will be "open". Such an ORF may contain a start codon and by definition cannot extend beyond a stop codon. That start codon indicates where translation may start. The transcription termination site is located after the ORF, beyond the translation stop codon. If transcription were to cease before the stop codon, an incomplete protein would be made during translation.
The completion of the human genome sequencing in the early 2000s was a turning point in genomics research. Scientists have conducted series of research into the activities of genes and the genome as a whole. The human genome contains around 3 billion base pairs nucleotide, and the huge quantity of data created necessitates the development of an accessible tool to explore and interpret this information in order to investigate the genetic basis of disease, evolution, and biological processes. The field of genomics has continued to grow, with new sequencing technologies and computational tool making it easier to study the genome.

A splice site mutation is a genetic mutation that inserts, deletes or changes a number of nucleotides in the specific site at which splicing takes place during the processing of precursor messenger RNA into mature messenger RNA. Splice site consensus sequences that drive exon recognition are located at the very termini of introns. The deletion of the splicing site results in one or more introns remaining in mature mRNA and may lead to the production of abnormal proteins. When a splice site mutation occurs, the mRNA transcript possesses information from these introns that normally should not be included. Introns are supposed to be removed, while the exons are expressed.

Transmembrane protein 50A is a protein that in humans is encoded by the TMEM50A gene.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
The Consensus Coding Sequence (CCDS) Project is a collaborative effort to maintain a dataset of protein-coding regions that are identically annotated on the human and mouse reference genome assemblies. The CCDS project tracks identical protein annotations on the reference mouse and human genomes with a stable identifier, and ensures that they are consistently represented by the National Center for Biotechnology Information (NCBI), Ensembl, and UCSC Genome Browser. The integrity of the CCDS dataset is maintained through stringent quality assurance testing and on-going manual curation.
Periannan Senapathy is a molecular biologist, geneticist, author and entrepreneur. He is the founder, president and chief scientific officer at Genome International Corporation, a biotechnology, bioinformatics, and information technology firm based in Madison, Wisconsin, which develops computational genomics applications of next-generation DNA sequencing (NGS) and clinical decision support systems for analyzing patient genome data that aids in diagnosis and treatment of diseases.
Christopher Boyce Burge is Professor of Biology and Biological Engineering at Massachusetts Institute of Technology.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
Chimeric RNA, sometimes referred to as a fusion transcript, is composed of exons from two or more different genes that have the potential to encode novel proteins. These mRNAs are different from those produced by conventional splicing as they are produced by two or more gene loci.
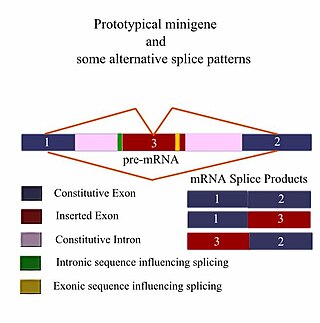
A minigene is a minimal gene fragment that includes an exon and the control regions necessary for the gene to express itself in the same way as a wild type gene fragment. This is a minigene in its most basic sense. More complex minigenes can be constructed containing multiple exons and intron(s). Minigenes provide a valuable tool for researchers evaluating splicing patterns both in vivo and in vitro biochemically assessed experiments. Specifically, minigenes are used as splice reporter vectors and act as a probe to determine which factors are important in splicing outcomes. They can be constructed to test the way both cis-regulatory elements and trans-regulatory elements affect gene expression.
WormBase is an online biological database about the biology and genome of the nematode model organism Caenorhabditis elegans and contains information about other related nematodes. WormBase is used by the C. elegans research community both as an information resource and as a place to publish and distribute their results. The database is regularly updated with new versions being released every two months. WormBase is one of the organizations participating in the Generic Model Organism Database (GMOD) project.
Single nucleotide polymorphism annotation is the process of predicting the effect or function of an individual SNP using SNP annotation tools. In SNP annotation the biological information is extracted, collected and displayed in a clear form amenable to query. SNP functional annotation is typically performed based on the available information on nucleic acid and protein sequences.

The Centre for Genomic Regulation is a biomedical and genomics research centre based on Barcelona. Most of its facilities and laboratories are located in the Barcelona Biomedical Research Park, in front of Somorrostro beach.
ANNOVAR is a bioinformatics software tool for the interpretation and prioritization of single nucleotide variants (SNVs), insertions, deletions, and copy number variants (CNVs) of a given genome.
References
- ↑ http://genes.mit.edu/GENSCAN.html Archived 2013-09-06 at the Wayback Machine The GENSCAN Web Server at MIT
- ↑ Burge, C. B. (1998) Modeling dependencies in pre-mRNA splicing signals. In Salzberg, S., Searls, D. and Kasif, S., eds. Computational Methods in Molecular Biology, Elsevier Science, Amsterdam, pp. 127-163. ISBN 978-0-444-50204-9
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Burge, Christopher; Karlin, Samuel (1997). "Prediction of complete gene structures in human genomic DNA" (PDF). Journal of Molecular Biology. 268 (1): 78–94. CiteSeerX 10.1.1.115.3107 . doi:10.1006/jmbi.1997.0951. PMID 9149143. Archived from the original (PDF) on 2015-06-20.
- ↑ Burge, C.; Karlin, S. (1998). "Finding the genes in genomic DNA". Current Opinion in Structural Biology. 8 (3): 346–354. doi: 10.1016/S0959-440X(98)80069-9 . PMID 9666331.
- 1 2 3 Flicek, Paul (2007). "Gene prediction: compare and CONTRAST". Genome Biology. 8 (12): 233. doi: 10.1186/gb-2007-8-12-233 . ISSN 1474-760X. PMC 2246255 . PMID 18096089.
- 1 2 Rogic, S.; Ouellette, B.F. F.; Mackworth, A. K. (2002-08-01). "Improving gene recognition accuracy by combining predictions from two gene-finding programs". Bioinformatics. 18 (8): 1034–1045. doi: 10.1093/bioinformatics/18.8.1034 . ISSN 1367-4803. PMID 12176826.