Glucocerebroside (also called glucosylceramide) is any of the cerebrosides in which the monosaccharide head group is glucose.
Contents


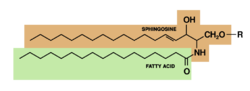
Research conducted on glucocerebrosides has shown that glucocerebrosides help support cellular functions in humans, such as signaling pathways as well as being possibly linked to diseases. Certain symptoms of diseases such as Gaucher's and Parkinson's disease have been linked to abnormal changes in glucocerebroside metabolism, such as changes in glucocerebroside levels and regulation. Researchers have also started to study the role of glucocerebrosides in cancer. [1]
In Gaucher's disease, the enzyme glucocerebrosidase is nonfunctional and cannot break down glucocerebroside into glucose and ceramide in the lysosome. [2] Affected macrophages, called Gaucher cells, have a distinct appearance similar to "wrinkled tissue paper" under light microscopy, because the substrates build-up within the lysosome. [3]
In 2019, research conducted by Lee et al., shows that glucocerebrosides impaired the formation of new blood vessels (angiogenesis) by decreasing the activity of Runx2 transcription factor, which leads to less vascular endothelial growth factor to be made. [4]
In 2023, research conducted by Russo et al., shows that increased levels of glucocerebroside can cause inflammation in dopamine-producing cells of the brain, which is usually seen in age-related diseases. The increased glucocerebroside levels is caused by a change in the SATB1-MIR22-GBA pathway, which leads to damage itolysosome and mitochondria lfunction. This damage is seen as a possible link to brain inflammation associated with Parkinson's disease. [5]