Muscular dystrophies (MD) are a genetically and clinically heterogeneous group of rare neuromuscular diseases that cause progressive weakness and breakdown of skeletal muscles over time. The disorders differ as to which muscles are primarily affected, the degree of weakness, how fast they worsen, and when symptoms begin. Some types are also associated with problems in other organs.

A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.

Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics. It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles. LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.

Duchenne muscular dystrophy (DMD) is a severe type of muscular dystrophy predominantly affecting boys. The onset of muscle weakness typically begins around age four, with rapid progression. Initially, muscle loss occurs in the thighs and pelvis, extending to the arms, which can lead to difficulties in standing up. By the age of 12, most individuals with Duchenne muscular dystrophy are unable to walk. Affected muscles may appear larger due to an increase in fat content, and scoliosis is common. Some individuals may experience intellectual disability, and females carrying a single copy of the mutated gene may show mild symptoms.

Becker muscular dystrophy (BMD) is an X-linked recessive inherited disorder characterized by slowly progressing muscle weakness of the legs and pelvis. It is a type of dystrophinopathy. The cause is mutations and deletions in any of the 79 exons encoding the large dystrophin protein, essential for maintaining the muscle fiber's cell membrane integrity. Becker muscular dystrophy is related to Duchenne muscular dystrophy in that both result from a mutation in the dystrophin gene, however the hallmark of Becker is milder in-frame deletions. and hence has a milder course, with patients maintaining ambulation till 50–60 years if detected early.

Oculopharyngeal muscular dystrophy (OPMD) is a rare form of muscular dystrophy with symptoms generally starting when an individual is 40 to 50 years old. It can be autosomal dominant neuromuscular disease or autosomal recessive. The most common inheritance of OPMD is autosomal dominant, which means only one copy of the mutated gene needs to be present in each cell. Children of an affected parent have a 50% chance of inheriting the mutant gene.

Fukuyama congenital muscular dystrophy (FCMD) is a rare, autosomal recessive form of muscular dystrophy (weakness and breakdown of muscular tissue) mainly described in Japan but also identified in Turkish and Ashkenazi Jewish patients; fifteen cases were first described on 1960 by Dr. Yukio Fukuyama.
In medicine, myopathy is a disease of the muscle in which the muscle fibers do not function properly. Myopathy means muscle disease. This meaning implies that the primary defect is within the muscle, as opposed to the nerves or elsewhere.
Nemaline myopathy is a congenital, often hereditary neuromuscular disorder with many symptoms that can occur such as muscle weakness, hypoventilation, swallowing dysfunction, and impaired speech ability. The severity of these symptoms varies and can change throughout one's life to some extent. The prevalence is estimated at 1 in 50,000 live births. It is the most common non-dystrophic myopathy.
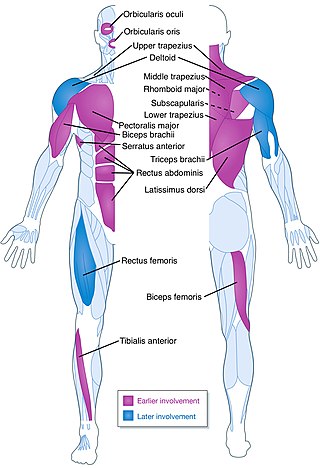
Facioscapulohumeral muscular dystrophy (FSHD) is a type of muscular dystrophy, a group of heritable diseases that cause degeneration of muscle and progressive weakness. Per the name, FSHD tends to sequentially weaken the muscles of the face, those that position the scapula, and those overlying the humerus bone of the upper arm. These areas can be spared, and muscles of other areas usually are affected, especially those of the chest, abdomen, spine, and shin. Almost any skeletal muscle can be affected in advanced disease. Abnormally positioned, termed 'winged', scapulas are common, as is the inability to lift the foot, known as foot drop. The two sides of the body are often affected unequally. Weakness typically manifests at ages 15–30 years. FSHD can also cause hearing loss and blood vessel abnormalities at the back of the eye.
Congenital muscular dystrophies are autosomal recessively-inherited muscle diseases. They are a group of heterogeneous disorders characterized by muscle weakness which is present at birth and the different changes on muscle biopsy that ranges from myopathic to overtly dystrophic due to the age at which the biopsy takes place.

Emery–Dreifuss muscular dystrophy (EDMD) is a type of muscular dystrophy, a group of heritable diseases that cause progressive impairment of muscles. EDMD affects muscles used for movement, causing atrophy, weakness and contractures. It almost always affects the heart, causing abnormal rhythms, heart failure, or sudden cardiac death. It is rare, affecting 0.39 per 100,000 people. It is named after Alan Eglin H. Emery and Fritz E. Dreifuss.
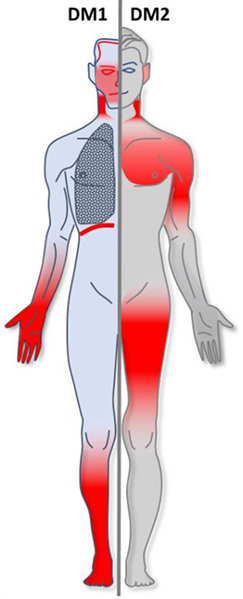
Myotonic dystrophy (DM) is a type of muscular dystrophy, a group of genetic disorders that cause progressive muscle loss and weakness. In DM, muscles are often unable to relax after contraction. Other manifestations may include cataracts, intellectual disability and heart conduction problems. In men, there may be early balding and infertility. While myotonic dystrophy can occur at any age, onset is typically in the 20s and 30s.
Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of limb-girdle muscular dystrophy. There are two types of Bethlem myopathy, based on which type of collagen is affected.

Choline kinase beta (CK), also known as Ethanolamine kinase (EK), Choline kinase-like protein , choline/ethanolamine kinase beta (CKEKB), or Choline/ethanolamine kinase is a protein encoded by the CHKB gene. This gene is found on chromosome 22 in humans. The encoded protein plays a key role in phospholipid biosynthesis. Choline kinase (CK) and ethanolamine kinase (EK) catalyzes the first step in phosphatidylethanolamine biosynthesis. Read-through transcripts are expressed from this locus that include exons from the downstream CPT1B locus.
Myopathic gait is a form of gait abnormality.
Ullrich congenital muscular dystrophy (UCMD) is a form of congenital muscular dystrophy. There are two forms: UCMD1 and UCMD2.
Acquired non-inflammatory myopathy (ANIM) is a neuromuscular disorder primarily affecting skeletal muscle, most commonly in the limbs of humans, resulting in a weakness or dysfunction in the muscle. A myopathy refers to a problem or abnormality with the myofibrils, which compose muscle tissue. In general, non-inflammatory myopathies are a grouping of muscular diseases not induced by an autoimmune-mediated inflammatory pathway. These muscular diseases usually arise from a pathology within the muscle tissue itself rather than the nerves innervating that tissue. ANIM has a wide spectrum of causes which include drugs and toxins, nutritional imbalances, acquired metabolic dysfunctions such as an acquired defect in protein structure, and infections.

Anoctamin 5 (ANO5) is a protein that in humans is encoded by the ANO5 gene.
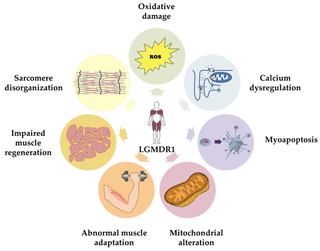
Calpainopathy is the most common type of autosomal recessive limb-girdle muscular dystrophy (LGMD). It preferentially affects the muscles of the hip girdle and shoulder girdle.