
ATPases (EC 3.6.1.3, adenylpyrophosphatase, ATP monophosphatase, triphosphatase, SV40 T-antigen, adenosine 5'-triphosphatase, ATP hydrolase, complex V (mitochondrial electron transport), (Ca2+ + Mg2+)-ATPase, HCO3−-ATPase, adenosine triphosphatase) are a class of enzymes that catalyze the decomposition of ATP into ADP and a free phosphate ion or the inverse reaction. This dephosphorylation reaction releases energy, which the enzyme (in most cases) harnesses to drive other chemical reactions that would not otherwise occur. This process is widely used in all known forms of life.

A transmembrane protein (TP) is a type of integral membrane protein that spans the entirety of the cell membrane. Many transmembrane proteins function as gateways to permit the transport of specific substances across the membrane. They frequently undergo significant conformational changes to move a substance through the membrane. They are usually highly hydrophobic and aggregate and precipitate in water. They require detergents or nonpolar solvents for extraction, although some of them (beta-barrels) can be also extracted using denaturing agents.
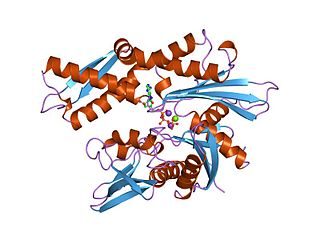
The 70 kilodalton heat shock proteins are a family of conserved ubiquitously expressed heat shock proteins. Proteins with similar structure exist in virtually all living organisms. The Hsp70s are an important part of the cell's machinery for protein folding, and help to protect cells from stress.

Hsp90 is a chaperone protein that assists other proteins to fold properly, stabilizes proteins against heat stress, and aids in protein degradation. It also stabilizes a number of proteins required for tumor growth, which is why Hsp90 inhibitors are investigated as anti-cancer drugs.

The Rossmann fold is a tertiary fold found in proteins that bind nucleotides, such as enzyme cofactors FAD, NAD+, and NADP+. This fold is composed of alternating beta strands and alpha helical segments where the beta strands are hydrogen bonded to each other forming an extended beta sheet and the alpha helices surround both faces of the sheet to produce a three-layered sandwich. The classical Rossmann fold contains six beta strands whereas Rossmann-like folds, sometimes referred to as Rossmannoid folds, contain only five strands. The initial beta-alpha-beta (bab) fold is the most conserved segment of the Rossmann fold. The motif is named after Michael Rossmann who first noticed this structural motif in the enzyme lactate dehydrogenase in 1970 and who later observed that this was a frequently occurring motif in nucleotide binding proteins.
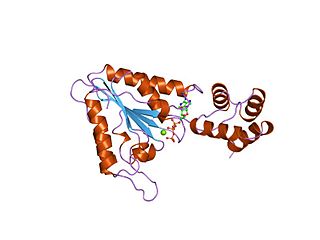
AAA proteins or ATPases Associated with diverse cellular Activities are a protein family sharing a common conserved module of approximately 230 amino acid residues. This is a large, functionally diverse protein family belonging to the AAA+ protein superfamily of ring-shaped P-loop NTPases, which exert their activity through the energy-dependent remodeling or translocation of macromolecules.

Menkes disease (MNK), also known as Menkes syndrome, is an X-linked recessive disorder caused by mutations in genes coding for the copper-transport protein ATP7A, leading to copper deficiency. Characteristic findings include kinky hair, growth failure, and nervous system deterioration. Like all X-linked recessive conditions, Menkes disease is more common in males than in females. The disorder was first described by John Hans Menkes in 1962.

The ATP-binding cassette transporters are a transport system superfamily that is one of the largest and possibly one of the oldest gene families. It is represented in all extant phyla, from prokaryotes to humans.

ATP7A, also known as Menkes' protein (MNK), is a copper-transporting P-type ATPase which uses the energy arising from ATP hydrolysis to transport Cu(I) across cell membranes. The ATP7A protein is a transmembrane protein and is expressed in the intestine and all tissues except liver. In the intestine, ATP7A regulates Cu(I) absorption in the human body by transporting Cu(I) from the small intestine into the blood. In other tissues, ATP7A shuttles between the Golgi apparatus and the cell membrane to maintain proper Cu(I) concentrations in the cell and provides certain enzymes with Cu(I). The X-linked, inherited, lethal genetic disorder of the ATP7A gene causes Menkes disease, a copper deficiency resulting in early childhood death.

In structural biology, a beta-propeller (β-propeller) is a type of all-β protein architecture characterized by 4 to 8 highly symmetrical blade-shaped beta sheets arranged toroidally around a central axis. Together the beta-sheets form a funnel-like active site.
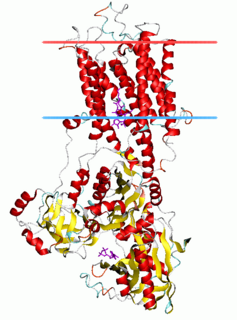
The P-type ATPases, also known as E1-E2 ATPases, are a large group of evolutionarily related ion and lipid pumps that are found in bacteria, archaea, and eukaryotes. P-type ATPases are α-helical bundle primary transporters named based upon their ability to catalyze auto- (or self-) phosphorylation (hence P) of a key conserved aspartate residue within the pump and their energy source, adenosine triphosphate (ATP). In addition, they all appear to interconvert between at least two different conformations, denoted by E1 and E2. P-type ATPases fall under the P-type ATPase (P-ATPase) Superfamily (TC# 3.A.3) which, as of early 2016, includes 20 different protein families.
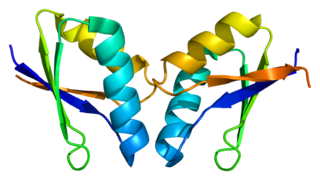
ATOX1 is a copper metallochaperone protein that is encoded by the ATOX1 gene in humans. In mammals, ATOX1 plays a key role in copper homeostasis as it delivers copper from the cytosol to transporters ATP7A and ATP7B. Homologous proteins are found in a wide variety of eukaryotes, including Saccharomyces cerevisiae as ATX1, and all contain a conserved metal binding domain.
The von Willebrand factor type A (vWA) domain is a protein domain named after its occurrence in von Willebrand factor (vWF), a large multimeric glycoprotein found in blood plasma. Mutant forms of vWF are involved in the aetiology of bleeding disorders. This type A domain is the prototype for a protein superfamily.
The Walker A and Walker B motifs are protein sequence motifs, known to have highly conserved three-dimensional structures. These were first reported in ATP-binding proteins by Walker and co-workers in 1982.

Copper is an essential trace element that is vital to the health of all living things. In humans, copper is essential to the proper functioning of organs and metabolic processes. The human body has complex homeostatic mechanisms which attempt to ensure a constant supply of available copper, while eliminating excess copper whenever this occurs. However, like all essential elements and nutrients, too much or too little nutritional ingestion of copper can result in a corresponding condition of copper excess or deficiency in the body, each of which has its own unique set of adverse health effects.

S-adenosylmethionine synthetase is an enzyme that creates S-adenosylmethionine by reacting methionine and ATP.
In molecular biology, the protein domain SAND is named after a range of proteins in the protein family: Sp100, AIRE-1, NucP41/75, DEAF-1. It is localised in the cell nucleus and has an important function in chromatin-dependent transcriptional control. It is found solely in eukaryotes.

In molecular biology, a carbohydrate-binding module (CBM) is a protein domain found in carbohydrate-active enzymes. The majority of these domains have carbohydrate-binding activity. Some of these domains are found on cellulosomal scaffoldin proteins. CBMs were previously known as cellulose-binding domains. CBMs are classified into numerous families, based on amino acid sequence similarity. There are currently 64 families of CBM in the CAZy database.

The mercury transporter superfamily is a family of transmembrane bacterial transporters of mercury ions. The common origin of all Mer superfamily members has been established. The common elements between family members are included in TMSs 1-2. A representative list of the subfamilies and proteins that belong to those subfamilies is available in the Transporter Classification Database.
Wilson disease protein (WND), also known as ATP7B protein, is a copper-transporting P-type ATPase which is encoded by the ATP7B gene. The ATP7B protein is located in the trans-Golgi network of the liver and brain and balances the copper level in the body by excreting excess copper into bile and plasma. Genetic disorder of the ATP7B gene may cause Wilson's disease, a disease in which copper accumulates in tissues, leading to neurological or psychiatric issues and liver diseases.
