Related Research Articles

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome that is present in a sufficiently large fraction of considered population.
A haplotype is a group of alleles in an organism that are inherited together from a single parent.
In population genetics, linkage disequilibrium (LD) is the non-random association of alleles at different loci in a given population. Loci are said to be in linkage disequilibrium when the frequency of association of their different alleles is higher or lower than expected if the loci were independent and associated randomly.
The International HapMap Project was an organization that aimed to develop a haplotype map (HapMap) of the human genome, to describe the common patterns of human genetic variation. HapMap is used to find genetic variants affecting health, disease and responses to drugs and environmental factors. The information produced by the project is made freely available for research.

An inversion is a chromosome rearrangement in which a segment of a chromosome becomes inverted within its original position. An inversion occurs when a chromosome undergoes a two breaks within the chromosomal arm, and the segment between the two breaks inserts itself in the opposite direction in the same chromosome arm. The breakpoints of inversions often happen in regions of repetitive nucleotides, and the regions may be reused in other inversions. Chromosomal segments in inversions can be as small as 100 kilobases or as large as 100 megabases. The number of genes captured by an inversion can range from a handful of genes to hundreds of genes. Inversions can happen either through ectopic recombination, chromosomal breakage and repair, or non-homologous end joining.

A DNA segment is identical by state (IBS) in two or more individuals if they have identical nucleotide sequences in this segment. An IBS segment is identical by descent (IBD) in two or more individuals if they have inherited it from a common ancestor without recombination, that is, the segment has the same ancestral origin in these individuals. DNA segments that are IBD are IBS per definition, but segments that are not IBD can still be IBS due to the same mutations in different individuals or recombinations that do not alter the segment.
Genetic association is when one or more genotypes within a population co-occur with a phenotypic trait more often than would be expected by chance occurrence.

In population genetics, an ancestry-informative marker (AIM) is a single-nucleotide polymorphism that exhibits substantially different frequencies between different populations. A set of many AIMs can be used to estimate the proportion of ancestry of an individual derived from each population.

Human genetic variation is the genetic differences in and among populations. There may be multiple variants of any given gene in the human population (alleles), a situation called polymorphism.
In molecular biology, SNP array is a type of DNA microarray which is used to detect polymorphisms within a population. A single nucleotide polymorphism (SNP), a variation at a single site in DNA, is the most frequent type of variation in the genome. Around 335 million SNPs have been identified in the human genome, 15 million of which are present at frequencies of 1% or higher across different populations worldwide.
A tag SNP is a representative single nucleotide polymorphism (SNP) in a region of the genome with high linkage disequilibrium that represents a group of SNPs called a haplotype. It is possible to identify genetic variation and association to phenotypes without genotyping every SNP in a chromosomal region. This reduces the expense and time of mapping genome areas associated with disease, since it eliminates the need to study every individual SNP. Tag SNPs are useful in whole-genome SNP association studies in which hundreds of thousands of SNPs across the entire genome are genotyped.

The Single Nucleotide Polymorphism Database (dbSNP) is a free public archive for genetic variation within and across different species developed and hosted by the National Center for Biotechnology Information (NCBI) in collaboration with the National Human Genome Research Institute (NHGRI). Although the name of the database implies a collection of one class of polymorphisms only, it in fact contains a range of molecular variation: (1) SNPs, (2) short deletion and insertion polymorphisms (indels/DIPs), (3) microsatellite markers or short tandem repeats (STRs), (4) multinucleotide polymorphisms (MNPs), (5) heterozygous sequences, and (6) named variants. The dbSNP accepts apparently neutral polymorphisms, polymorphisms corresponding to known phenotypes, and regions of no variation. It was created in September 1998 to supplement GenBank, NCBI’s collection of publicly available nucleic acid and protein sequences.
In genetics, association mapping, also known as "linkage disequilibrium mapping", is a method of mapping quantitative trait loci (QTLs) that takes advantage of historic linkage disequilibrium to link phenotypes to genotypes, uncovering genetic associations.
Nested association mapping (NAM) is a technique designed by the labs of Edward Buckler, James Holland, and Michael McMullen for identifying and dissecting the genetic architecture of complex traits in corn. It is important to note that nested association mapping is a specific technique that cannot be performed outside of a specifically designed population such as the Maize NAM population, the details of which are described below.
Genomic structural variation is the variation in structure of an organism's chromosome. It consists of many kinds of variation in the genome of one species, and usually includes microscopic and submicroscopic types, such as deletions, duplications, copy-number variants, insertions, inversions and translocations. Originally, a structure variation affects a sequence length about 1kb to 3Mb, which is larger than SNPs and smaller than chromosome abnormality. However, the operational range of structural variants has widened to include events > 50bp. The definition of structural variation does not imply anything about frequency or phenotypical effects. Many structural variants are associated with genetic diseases, however many are not. Recent research about SVs indicates that SVs are more difficult to detect than SNPs. Approximately 13% of the human genome is defined as structurally variant in the normal population, and there are at least 240 genes that exist as homozygous deletion polymorphisms in human populations, suggesting these genes are dispensable in humans. Rapidly accumulating evidence indicates that structural variations can comprise millions of nucleotides of heterogeneity within every genome, and are likely to make an important contribution to human diversity and disease susceptibility.
The Functional Element SNPs Database (FESD) is a biological database of single nucleotide polymorphisms in molecular biology. The database is a tool designed to organize functional elements into categories in human gene regions and to output their sequences needed for genotyping experiments as well as provide a set of SNPs that lie within each region. The database defines functional elements into ten types: promoter regions, CpG islands,5' untranslated regions (5'-UTRs), translation start sites, splice sites, coding exons, introns, translation stop sites, polyadenylation signals, and 3' UTRs. People may reference this database for haplotype information or obtain a flanking sequence for genotyping. This may help in finding mutations that contribute to common and polygenic diseases. Researchers can manually choose a group of SNPs of special interest for certain functional elements along with their corresponding sequences. The database combines information from sources such as HapMap, UCSC GoldenPath, dbSNP, OMIM, and TRANSFAC. Users can obtain information about tag SNPs and simulate LD blocks for each gene. FESD is still a developing database and is not widely known so was unable to find projects that used the database. Research was found using similar databases or databases that are combined in FESD's information pool.

PR domain zinc finger protein 9 is a protein that in humans is encoded by the PRDM9 gene. PRDM9 is responsible for positioning recombination hotspots during meiosis by binding a DNA sequence motif encoded in its zinc finger domain. PRDM9 is the only speciation gene found so far in mammals, and is one of the fastest evolving genes in the genome.

David Matthew Altshuler is a clinical endocrinologist and human geneticist. He is Executive Vice President, Global Research and Chief Scientific Officer at Vertex Pharmaceuticals. Prior to joining Vertex in 2014, he was at the Broad Institute of Harvard and MIT, and was a Professor of Genetics and Medicine at Harvard Medical School, and in the Department of Biology at Massachusetts Institute of Technology. He was also a faculty member in the Department of Molecular Biology, Center for Human Genetic Research, and the Diabetes Unit, all at Massachusetts General Hospital. He was one of four Founding Core Members of the Broad Institute, and served as the Institute's Deputy Director, Chief Academic Officer, and Director of the Program in Medical and Population Genetics.
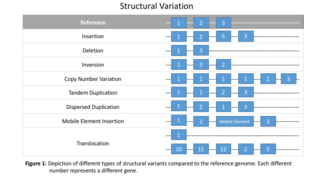
Structural variation in the human genome is operationally defined as genomic alterations, varying between individuals, that involve DNA segments larger than 1 kilo base (kb), and could be either microscopic or submicroscopic. This definition distinguishes them from smaller variants that are less than 1 kb in size such as short deletions, insertions, and single nucleotide variants.
Amanda M. Hulse-Kemp is a computational biologist with the United States Department of Agriculture – Agricultural Research Service. She works in the Genomics and Bioinformatics Research Unit and is stationed on the North Carolina State University campus in Raleigh, North Carolina.
References
- ↑ Gabriel, S. B.; Daly, Mark J.; Lander, Eric S.; Ward, Ryk; Cooper, Richard; Adeyemo, Adebowale; Rotimi, Charles; Liu-Cordero, Shau Neen; Faggart, Maura (2002-06-21). "The Structure of Haplotype Blocks in the Human Genome". Science . 296 (5576): 2225–2229. Bibcode:2002Sci...296.2225G. doi: 10.1126/science.1069424 . ISSN 0036-8075. PMID 12029063. S2CID 10069634.
- ↑ Wall, Jeffrey D.; Pritchard, Jonathan K. (1 August 2003). "Haplotype blocks and linkage disequilibrium in the human genome". Nature Reviews Genetics . 4 (8): 587–597. doi:10.1038/nrg1123. PMID 12897771. S2CID 15478115.
- ↑ Schulze, Thomas G.; Zhang, Kui; Chen, Yu-Sheng; Akula, Nirmala; Sun, Fengzhu; McMahon, Francis J. (1 February 2004). "Defining haplotype blocks and tag single-nucleotide polymorphisms in the human genome". Human Molecular Genetics . 13 (3): 335–342. doi: 10.1093/hmg/ddh035 . PMID 14681300.
- ↑ Indap, Amit R; Marth, Gabor T; Struble, Craig A; Tonellato, Peter; Olivier, Michael (2005). "Analysis of concordance of different haplotype block partitioning algorithms". BMC Bioinformatics . 6 (1): 303. doi: 10.1186/1471-2105-6-303 . PMC 1343594 . PMID 16356172.
- ↑ Cardon, Lon R.; Abecasis, Gonçalo R. (March 2003). "Using haplotype blocks to map human complex trait loci". Trends in Genetics . 19 (3): 135–140. doi:10.1016/S0168-9525(03)00022-2. PMID 12615007.
- ↑ Wang, Ning; Akey, Joshua M.; Zhang, Kun; Chakraborty, Ranajit; Jin, Li (November 2002). "Distribution of Recombination Crossovers and the Origin of Haplotype Blocks: The Interplay of Population History, Recombination, and Mutation". The American Journal of Human Genetics . 71 (5): 1227–1234. doi:10.1086/344398. PMC 385104 . PMID 12384857.
- ↑ Patil, N; Berno, AJ; Hinds, DA; Barrett, WA; Doshi, JM; Hacker, CR; Kautzer, CR; Lee, DH; Marjoribanks, C; McDonough, DP; Nguyen, BT; Norris, MC; Sheehan, JB; Shen, N; Stern, D; Stokowski, RP; Thomas, DJ; Trulson, MO; Vyas, KR; Frazer, KA; Fodor, SP; Cox, DR (23 November 2001). "Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21". Science. 294 (5547): 1719–23. Bibcode:2001Sci...294.1719P. doi:10.1126/science.1065573. PMID 11721056. S2CID 14168219.
- ↑ Zhang, Kui; Calabrese, Peter; Nordborg, Magnus; Sun, Fengzhu (December 2002). "Haplotype Block Structure and Its Applications to Association Studies: Power and Study Designs". The American Journal of Human Genetics. 71 (6): 1386–1394. doi:10.1086/344780. PMC 378580 . PMID 12439824.
| | This genetics article is a stub. You can help Wikipedia by expanding it. |