Related Research Articles

The proteome is the entire set of proteins that is, or can be, expressed by a genome, cell, tissue, or organism at a certain time. It is the set of expressed proteins in a given type of cell or organism, at a given time, under defined conditions. Proteomics is the study of the proteome.

Proteomics is the large-scale study of proteins. Proteins are vital parts of living organisms, with many functions such as the formation of structural fibers of muscle tissue, enzymatic digestion of food, or synthesis and replication of DNA. In addition, other kinds of proteins include antibodies that protect an organism from infection, and hormones that send important signals throughout the body.
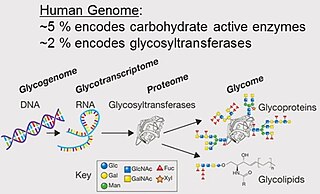
A glycome is the entire complement or complete set of all sugars, whether free or chemically bound in more complex molecules, of an organism. An alternative definition is the entirety of carbohydrates in a cell. The glycome may in fact be one of the most complex entities in nature. "Glycomics, analogous to genomics and proteomics, is the systematic study of all glycan structures of a given cell type or organism" and is a subset of glycobiology.

Two-dimensional gel electrophoresis, abbreviated as 2-DE or 2-D electrophoresis, is a form of gel electrophoresis commonly used to analyze proteins. Mixtures of proteins are separated by two properties in two dimensions on 2D gels. 2-DE was first independently introduced by O'Farrell and Klose in 1975.
Blood-proteins, also termed plasma proteins, are proteins present in blood plasma. They serve many different functions, including transport of lipids, hormones, vitamins and minerals in activity and functioning of the immune system. Other blood proteins act as enzymes, complement components, protease inhibitors or kinin precursors. Contrary to popular belief, haemoglobin is not a blood protein, as it is carried within red blood cells, rather than in the blood serum.

Peptide mass fingerprinting (PMF), also known as protein fingerprinting, is an analytical technique for protein identification in which the unknown protein of interest is first cleaved into smaller peptides, whose absolute masses can be accurately measured with a mass spectrometer such as MALDI-TOF or ESI-TOF. The method was developed in 1993 by several groups independently. The peptide masses are compared to either a database containing known protein sequences or even the genome. This is achieved by using computer programs that translate the known genome of the organism into proteins, then theoretically cut the proteins into peptides, and calculate the absolute masses of the peptides from each protein. They then compare the masses of the peptides of the unknown protein to the theoretical peptide masses of each protein encoded in the genome. The results are statistically analyzed to find the best match.
QPNC-PAGE, or QuantitativePreparativeNativeContinuousPolyacrylamideGel Electrophoresis, is a bioanalytical, one-dimensional, high-resolution and high-precision electrophoresis technique applied in biochemistry and bioinorganic chemistry to separate proteins quantitatively by isoelectric point and by continuous elution from a gel column.
Surface-enhanced laser desorption/ionization (SELDI) is a soft ionization method in mass spectrometry (MS) used for the analysis of protein mixtures. It is a variation of matrix-assisted laser desorption/ionization (MALDI). In MALDI, the sample is mixed with a matrix material and applied to a metal plate before irradiation by a laser, whereas in SELDI, proteins of interest in a sample become bound to a surface before MS analysis. The sample surface is a key component in the purification, desorption, and ionization of the sample. SELDI is typically used with time-of-flight (TOF) mass spectrometers and is used to detect proteins in tissue samples, blood, urine, or other clinical samples, however, SELDI technology can potentially be used in any application by simply modifying the sample surface.

MALDI mass spectrometry imaging (MALDI-MSI) is the use of matrix-assisted laser desorption ionization as a mass spectrometry imaging technique in which the sample, often a thin tissue section, is moved in two dimensions while the mass spectrum is recorded. Advantages, like measuring the distribution of a large amount of analytes at one time without destroying the sample, make it a useful method in tissue-based study.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.
Shotgun proteomics refers to the use of bottom-up proteomics techniques in identifying proteins in complex mixtures using a combination of high performance liquid chromatography combined with mass spectrometry. The name is derived from shotgun sequencing of DNA which is itself named after the rapidly expanding, quasi-random firing pattern of a shotgun. The most common method of shotgun proteomics starts with the proteins in the mixture being digested and the resulting peptides are separated by liquid chromatography. Tandem mass spectrometry is then used to identify the peptides.
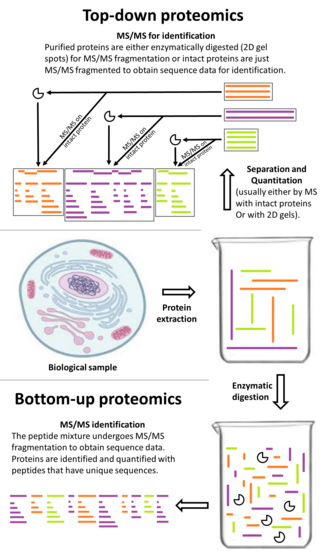
Top-down proteomics is a method of protein identification that either uses an ion trapping mass spectrometer to store an isolated protein ion for mass measurement and tandem mass spectrometry (MS/MS) analysis or other protein purification methods such as two-dimensional gel electrophoresis in conjunction with MS/MS. Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. The name is derived from the similar approach to DNA sequencing. During mass spectrometry intact proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance, quadrupole ion trap or Orbitrap mass spectrometer. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. Effective fractionation is critical for sample handling before mass-spectrometry-based proteomics. Proteome analysis routinely involves digesting intact proteins followed by inferred protein identification using mass spectrometry (MS). Top-down MS (non-gel) proteomics interrogates protein structure through measurement of an intact mass followed by direct ion dissociation in the gas phase.
Bottom-up proteomics is a common method to identify proteins and characterize their amino acid sequences and post-translational modifications by proteolytic digestion of proteins prior to analysis by mass spectrometry. The major alternative workflow used in proteomics is called top-down proteomics where intact proteins are purified prior to digestion and/or fragmentation either within the mass spectrometer or by 2D electrophoresis. Essentially, bottom-up proteomics is a relatively simple and reliable means of determining the protein make-up of a given sample of cells, tissues, etc.

Quantitative proteomics is an analytical chemistry technique for determining the amount of proteins in a sample. The methods for protein identification are identical to those used in general proteomics, but include quantification as an additional dimension. Rather than just providing lists of proteins identified in a certain sample, quantitative proteomics yields information about the physiological differences between two biological samples. For example, this approach can be used to compare samples from healthy and diseased patients. Quantitative proteomics is mainly performed by two-dimensional gel electrophoresis (2-DE), preparative native PAGE, or mass spectrometry (MS). However, a recent developed method of quantitative dot blot (QDB) analysis is able to measure both the absolute and relative quantity of an individual proteins in the sample in high throughput format, thus open a new direction for proteomic research. In contrast to 2-DE, which requires MS for the downstream protein identification, MS technology can identify and quantify the changes.

Elongation factor Tu, mitochondrial is a protein that in humans is encoded by the TUFM gene. It is an EF-Tu homolog.
The Proteomics Standards Initiative (PSI) is a working group of the Human Proteome Organization. It aims to define data standards for proteomics to facilitate data comparison, exchange and verification.

Capillary electrophoresis–mass spectrometry (CE–MS) is an analytical chemistry technique formed by the combination of the liquid separation process of capillary electrophoresis with mass spectrometry. CE–MS combines advantages of both CE and MS to provide high separation efficiency and molecular mass information in a single analysis. It has high resolving power and sensitivity, requires minimal volume and can analyze at high speed. Ions are typically formed by electrospray ionization, but they can also be formed by matrix-assisted laser desorption/ionization or other ionization techniques. It has applications in basic research in proteomics and quantitative analysis of biomolecules as well as in clinical medicine. Since its introduction in 1987, new developments and applications have made CE-MS a powerful separation and identification technique. Use of CE–MS has increased for protein and peptides analysis and other biomolecules. However, the development of online CE–MS is not without challenges. Understanding of CE, the interface setup, ionization technique and mass detection system is important to tackle problems while coupling capillary electrophoresis to mass spectrometry.

Affinity electrophoresis is a general name for many analytical methods used in biochemistry and biotechnology. Both qualitative and quantitative information may be obtained through affinity electrophoresis. Cross electrophoresis, the first affinity electrophoresis method, was created by Nakamura et al. Enzyme-substrate complexes have been detected using cross electrophoresis. The methods include the so-called electrophoretic mobility shift assay, charge shift electrophoresis and affinity capillary electrophoresis. The methods are based on changes in the electrophoretic pattern of molecules through biospecific interaction or complex formation. The interaction or binding of a molecule, charged or uncharged, will normally change the electrophoretic properties of a molecule. Membrane proteins may be identified by a shift in mobility induced by a charged detergent. Nucleic acids or nucleic acid fragments may be characterized by their affinity to other molecules. The methods have been used for estimation of binding constants, as for instance in lectin affinity electrophoresis or characterization of molecules with specific features like glycan content or ligand binding. For enzymes and other ligand-binding proteins, one-dimensional electrophoresis similar to counter electrophoresis or to "rocket immunoelectrophoresis", affinity electrophoresis may be used as an alternative quantification of the protein. Some of the methods are similar to affinity chromatography by use of immobilized ligands.

In the field of cellular biology, single-cell analysis and subcellular analysis is the study of genomics, transcriptomics, proteomics, metabolomics and cell–cell interactions at the single cell level. The concept of single-cell analysis originated in the 1970s. Before the discovery of heterogeneity, single-cell analysis mainly referred to the analysis or manipulation of an individual cell in a bulk population of cells at a particular condition using optical or electronic microscope. To date, due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells. Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies, enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome and transcriptome of single cells, as well as to quantify their proteome and metabolome. Mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells. Recent advances have enabled quantifying thousands of protein across hundreds of single cells, and thus make possible new types of analysis. In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.

Ancient proteins are complex mixtures and the term palaeoproteomics is used to characterise the study of proteomes in the past. Ancients proteins have been recovered from a wide range of archaeological materials, including bones, teeth, eggshells, leathers, parchments, ceramics, painting binders and well-preserved soft tissues like gut intestines. These preserved proteins have provided valuable information about taxonomic identification, evolution history (phylogeny), diet, health, disease, technology and social dynamics in the past.
References
- ↑ Svensson M, et al. (February 2009). "Heat Stabilization of the Tissue Proteome: A New Technology for Improved Proteomics". J. Proteome Res. 8 (2): 974–981. CiteSeerX 10.1.1.464.2789 . doi:10.1021/pr8006446. PMID 19159280.
- ↑ Sköld K, Alm H, Scholz B (June 2013). "The impact of biosampling procedures on molecular data interpretation". Mol. Cell. Proteomics (Review). 12 (6): 1489–1501. doi: 10.1074/mcp.R112.024869 . PMC 3675808 . PMID 23382104.
- ↑ Söderquist M (15 January 2013). "Eliminating biological change after excision". Gen. Eng. Biotechnol. News (Tutorial). 33 (2).
- ↑ Smejkal GB, et al. (August 2011). "Thermal stabilization of tissues and the preservation of protein phosphorylation states for two-dimensional gel electrophoresis". Electrophoresis. 32 (16): 2206–2215. doi:10.1002/elps.201100170. PMID 21792998.
- ↑ Lundby A, et al. (June 2012). "Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues". Nat. Commun. 3 (876): 876. Bibcode:2012NatCo...3..876L. doi:10.1038/ncomms1871. PMC 3621391 . PMID 22673903.
- ↑ Blatherwick EQ, et al. (July 2013). "Localisation of adenine nucleotides in heat-stabilised mouse brains using ion mobility enabled MALDI Imaging". Int. J. Mass Spectrom. 345–347: 19–27. Bibcode:2013IJMSp.345...19B. doi:10.1016/j.ijms.2013.02.004.
- ↑ Spellman C, et al. (January 2013). "Expression of trisomic proteins in Down syndrome model systems". Gene. 512 (2): 219–225. doi:10.1016/j.gene.2012.10.051. PMID 23103828.
- ↑ Ahmed MM, et al. (May 2013). "Protein profiles in Tc1 mice implicate novel pathway perturbations in the Down syndrome brain". Hum. Mol. Genet. 22 (9): 1709–1724. doi:10.1093/hmg/ddt017. PMC 3613160 . PMID 23349361.
- ↑ "CDC Invests in Denator's Heat Stabilization Technology". News: Products & Services. Gen. Eng. Biotechnol. News. 36 (14): 8. August 2016.