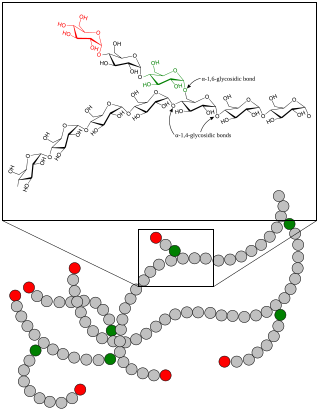
A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.

Hyperammonemia is a metabolic disturbance characterised by an excess of ammonia in the blood. It is a dangerous condition that may lead to brain injury and death. It may be primary or secondary.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are now often referred to as congenital metabolic diseases or inherited metabolic disorders. To this concept it's possible to include the new term of Enzymopathy. This term was created following the study of Biodynamic Enzymology, a science based on the study of the enzymes and their derivated products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene-one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.
Charles Robert Scriver was a Canadian pediatrician and biochemical geneticist. His work focused on inborn errors of metabolism and led in establishing a Canada-wide newborn metabolic screening program.

Sir Archibald Edward Garrod was an English physician who pioneered the field of inborn errors of metabolism. He also discovered alkaptonuria, understanding its inheritance. He served as Regius Professor of Medicine at the University of Oxford from 1920 to 1927.

A metabolic disorder is a disorder that negatively alters the body's processing and distribution of macronutrients, such as proteins, fats, and carbohydrates. Metabolic disorders can happen when abnormal chemical reactions in the body alter the normal metabolic process. It can also be defined as inherited single gene anomaly, most of which are autosomal recessive.
Disaccharidases are glycoside hydrolases, enzymes that break down certain types of sugars called disaccharides into simpler sugars called monosaccharides. In the human body, disaccharidases are made mostly in an area of the small intestine's wall called the brush border, making them members of the group of "brush border enzymes".

Medical genetics is the branch of medicine that involves the diagnosis and management of hereditary disorders. Medical genetics differs from human genetics in that human genetics is a field of scientific research that may or may not apply to medicine, while medical genetics refers to the application of genetics to medical care. For example, research on the causes and inheritance of genetic disorders would be considered within both human genetics and medical genetics, while the diagnosis, management, and counselling people with genetic disorders would be considered part of medical genetics.

Glycine encephalopathy is a rare autosomal recessive disorder of glycine metabolism. After phenylketonuria, glycine encephalopathy is the second most common disorder of amino acid metabolism. The disease is caused by defects in the glycine cleavage system, an enzyme responsible for glycine catabolism. There are several forms of the disease, with varying severity of symptoms and time of onset. The symptoms are exclusively neurological in nature, and clinically this disorder is characterized by abnormally high levels of the amino acid glycine in bodily fluids and tissues, especially the cerebrospinal fluid.
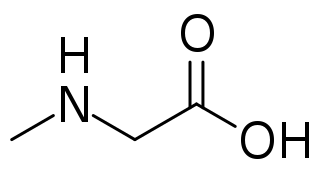
Sarcosinemia (SAR), also called hypersarcosinemia and SARDH deficiency, is a rare autosomal recessive metabolic disorder characterized by an increased concentration of sarcosine in blood plasma and urine ("sarcosinuria"). It can result from an inborn error of sarcosine metabolism, or from severe folate deficiency related to the folate requirement for the conversion of sarcosine to glycine. It is thought to be a relatively benign condition.
Metab-L is an electronic mailing list on inborn errors of metabolism (IEM) that has acquired notability among specialists in that field of medicine, especially from the area of pediatrics.

Essential fructosuria, caused by a deficiency of the enzyme hepatic fructokinase, is a clinically benign condition characterized by the incomplete metabolism of fructose in the liver, leading to its excretion in urine. Fructokinase is the first enzyme involved in the degradation of fructose to fructose-1-phosphate in the liver.
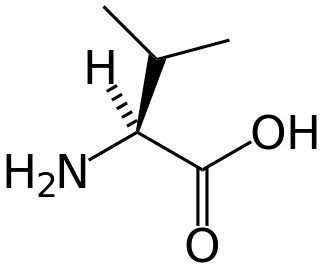
Hypervalinemia is a rare autosomal recessive metabolic disorder in which urinary and serum levels of the branched-chain amino acid valine are elevated, without related elevation of the branched-chain amino acids leucine and isoleucine. It is caused by a deficiency of the enzyme valine transaminase.

Urocanic aciduria is an autosomal recessive metabolic disorder caused by a deficiency of the enzyme urocanase. It is a secondary disorder of histidine metabolism.

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.

Succinyl-CoA:3-oxoacid CoA transferase deficiency is an inborn error of ketone body utilization. Succinyl-CoA:3-oxoacid CoA transferase catalyzes the transfer of coenzyme A from succinyl-coenzyme A to acetoacetate. It can be caused by mutation in the OXCT1 gene.
Metabolic myopathies are myopathies that result from defects in biochemical metabolism that primarily affect muscle. They are generally genetic defects that interfere with muscle's ability to create energy, causing a low ATP reservoir within the muscle cell.
In medicine, Garrod's tetrad is a term named for British physician Archibald Garrod, who introduced the phrase "inborn errors of metabolism" in a lecture in 1908.
D-Glyceric Acidemia is an inherited disease, in the category of inborn errors of metabolism. It is caused by a mutation in the gene GLYCTK, which encodes for the enzyme glycerate kinase.
Harvey Louis Levy is an American biochemical geneticist, pediatrician, physician scientist and academic. He is Senior Physician in Medicine and Genetics at Boston Children’s Hospital and Professor of Pediatrics at Harvard Medical School.