In genetics, shotgun sequencing is a method used for sequencing random DNA strands. It is named by analogy with the rapidly expanding, quasi-random shot grouping of a shotgun.
A bacterial artificial chromosome (BAC) is a DNA construct, based on a functional fertility plasmid, used for transforming and cloning in bacteria, usually E. coli. F-plasmids play a crucial role because they contain partition genes that promote the even distribution of plasmids after bacterial cell division. The bacterial artificial chromosome's usual insert size is 150–350 kbp. A similar cloning vector called a PAC has also been produced from the DNA of P1 bacteriophage.
A contig is a set of overlapping DNA segments that together represent a consensus region of DNA. In bottom-up sequencing projects, a contig refers to overlapping sequence data (reads); in top-down sequencing projects, contig refers to the overlapping clones that form a physical map of the genome that is used to guide sequencing and assembly. Contigs can thus refer both to overlapping DNA sequences and to overlapping physical segments (fragments) contained in clones depending on the context.

Yeast artificial chromosomes (YACs) are genetically engineered chromosomes derived from the DNA of the yeast, Saccharomyces cerevisiae, which is then ligated into a bacterial plasmid. By inserting large fragments of DNA, from 100–1000 kb, the inserted sequences can be cloned and physically mapped using a process called chromosome walking. This is the process that was initially used for the Human Genome Project, however due to stability issues, YACs were abandoned for the use of bacterial artificial chromosome
Chromosome jumping is a tool of molecular biology that is used in the physical mapping of genomes. It is related to several other tools used for the same purpose, including chromosome walking.
Primer walking is a technique used to clone a gene from its known closest markers. As a result, it is employed in cloning and sequencing efforts in plants, fungi, and mammals with minor alterations. This technique, also known as "directed sequencing," employs a series of Sanger sequencing reactions to either confirm the reference sequence of a known plasmid or PCR product based on the reference sequence or to discover the unknown sequence of a full plasmid or PCR product by designing primers to sequence overlapping sections.
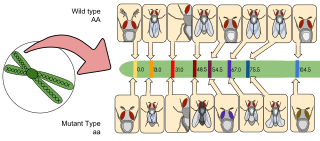
Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.
A genomic library is a collection of overlapping DNA fragments that together make up the total genomic DNA of a single organism. The DNA is stored in a population of identical vectors, each containing a different insert of DNA. In order to construct a genomic library, the organism's DNA is extracted from cells and then digested with a restriction enzyme to cut the DNA into fragments of a specific size. The fragments are then inserted into the vector using DNA ligase. Next, the vector DNA can be taken up by a host organism - commonly a population of Escherichia coli or yeast - with each cell containing only one vector molecule. Using a host cell to carry the vector allows for easy amplification and retrieval of specific clones from the library for analysis.
Fosmids are similar to cosmids but are based on the bacterial F-plasmid. The cloning vector is limited, as a host can only contain one fosmid molecule. Fosmids can hold DNA inserts of up to 40 kb in size; often the source of the insert is random genomic DNA. A fosmid library is prepared by extracting the genomic DNA from the target organism and cloning it into the fosmid vector. The ligation mix is then packaged into phage particles and the DNA is transfected into the bacterial host. Bacterial clones propagate the fosmid library. The low copy number offers higher stability than vectors with relatively higher copy numbers, including cosmids. Fosmids may be useful for constructing stable libraries from complex genomes. Fosmids have high structural stability and have been found to maintain human DNA effectively even after 100 generations of bacterial growth. Fosmid clones were used to help assess the accuracy of the Public Human Genome Sequence.
Paired-end tags (PET) are the short sequences at the 5’ and 3' ends of a DNA fragment which are unique enough that they (theoretically) exist together only once in a genome, therefore making the sequence of the DNA in between them available upon search or upon further sequencing. Paired-end tags (PET) exist in PET libraries with the intervening DNA absent, that is, a PET "represents" a larger fragment of genomic or cDNA by consisting of a short 5' linker sequence, a short 5' sequence tag, a short 3' sequence tag, and a short 3' linker sequence. It was shown conceptually that 13 base pairs are sufficient to map tags uniquely. However, longer sequences are more practical for mapping reads uniquely. The endonucleases used to produce PETs give longer tags but sequences of 50–100 base pairs would be optimal for both mapping and cost efficiency. After extracting the PETs from many DNA fragments, they are linked (concatenated) together for efficient sequencing. On average, 20–30 tags could be sequenced with the Sanger method, which has a longer read length. Since the tag sequences are short, individual PETs are well suited for next-generation sequencing that has short read lengths and higher throughput. The main advantages of PET sequencing are its reduced cost by sequencing only short fragments, detection of structural variants in the genome, and increased specificity when aligning back to the genome compared to single tags, which involves only one end of the DNA fragment.
Optical mapping is a technique for constructing ordered, genome-wide, high-resolution restriction maps from single, stained molecules of DNA, called "optical maps". By mapping the location of restriction enzyme sites along the unknown DNA of an organism, the spectrum of resulting DNA fragments collectively serves as a unique "fingerprint" or "barcode" for that sequence. Originally developed by Dr. David C. Schwartz and his lab at NYU in the 1990s this method has since been integral to the assembly process of many large-scale sequencing projects for both microbial and eukaryotic genomes. Later technologies use DNA melting, DNA competitive binding or enzymatic labelling in order to create the optical mappings.
Polony sequencing is an inexpensive but highly accurate multiplex sequencing technique that can be used to “read” millions of immobilized DNA sequences in parallel. This technique was first developed by Dr. George Church's group at Harvard Medical School. Unlike other sequencing techniques, Polony sequencing technology is an open platform with freely downloadable, open source software and protocols. Also, the hardware of this technique can be easily set up with a commonly available epifluorescence microscopy and a computer-controlled flowcell/fluidics system. Polony sequencing is generally performed on paired-end tags library that each molecule of DNA template is of 135 bp in length with two 17–18 bp paired genomic tags separated and flanked by common sequences. The current read length of this technique is 26 bases per amplicon and 13 bases per tag, leaving a gap of 4–5 bases in each tag.
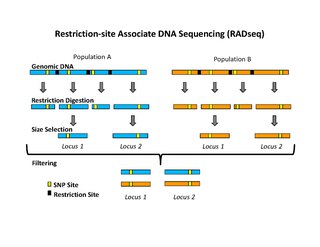
Restriction site associated DNA (RAD) markers are a type of genetic marker which are useful for association mapping, QTL-mapping, population genetics, ecological genetics and evolutionary genetics. The use of RAD markers for genetic mapping is often called RAD mapping. An important aspect of RAD markers and mapping is the process of isolating RAD tags, which are the DNA sequences that immediately flank each instance of a particular restriction site of a restriction enzyme throughout the genome. Once RAD tags have been isolated, they can be used to identify and genotype DNA sequence polymorphisms mainly in form of single nucleotide polymorphisms (SNPs). Polymorphisms that are identified and genotyped by isolating and analyzing RAD tags are referred to as RAD markers. Although genotyping by sequencing presents an approach similar to the RAD-seq method, they differ in some substantial ways.
In DNA sequencing, a read is an inferred sequence of base pairs corresponding to all or part of a single DNA fragment. A typical sequencing experiment involves fragmentation of the genome into millions of molecules, which are size-selected and ligated to adapters. The set of fragments is referred to as a sequencing library, which is sequenced to produce a set of reads.

End-sequence profiling (ESP) is a method based on sequence-tagged connectors developed to facilitate de novo genome sequencing to identify high-resolution copy number and structural aberrations such as inversions and translocations.
A plant genome assembly represents the complete genomic sequence of a plant species, which is assembled into chromosomes and other organelles by using DNA fragments that are obtained from different types of sequencing technology.
BLESS, also known as breaks labeling, enrichment on streptavidin and next-generation sequencing, is a method used to detect genome-wide double-strand DNA damage. In contrast to chromatin immunoprecipitation (ChIP)-based methods of identifying DNA double-strand breaks (DSBs) by labeling DNA repair proteins, BLESS utilizes biotinylated DNA linkers to directly label genomic DNA in situ which allows for high-specificity enrichment of samples on streptavidin beads and the subsequent sequencing-based DSB mapping to nucleotide resolution.
Physical map is a technique used in molecular biology to find the order and physical distance between DNA base pairs by DNA markers. It is one of the gene mapping techniques which can determine the sequence of DNA base pairs with high accuracy. Genetic mapping, another approach of gene mapping, can provide markers needed for the physical mapping. However, as the former deduces the relative gene position by recombination frequencies, it is less accurate than the latter.

Hi-C is a high-throughput genomic and epigenomic technique to capture chromatin conformation. In general, Hi-C is considered as a derivative of a series of chromosome conformation capture technologies, including but not limited to 3C, 4C, and 5C. Hi-C comprehensively detects genome-wide chromatin interactions in the cell nucleus by combining 3C and next-generation sequencing (NGS) approaches and has been considered as a qualitative leap in C-technology development and the beginning of 3D genomics.
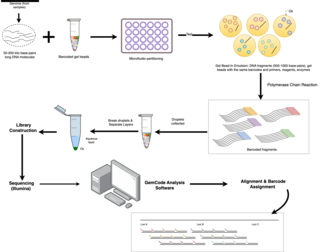
Linked-read sequencing, a type of DNA sequencing technology, uses specialized technique that tags DNA molecules with unique barcodes before fragmenting them. Unlike traditional sequencing technology, where DNA is broken into small fragments and then sequenced individually, resulting in short read lengths that has difficulties in accurately reconstructing the original DNA sequence, the unique barcodes of linked-read sequencing allows scientists to link together DNA fragments that come from the same DNA molecule. A pivotal benefit of this technology lies in the small quantities of DNA required for large genome information output, effectively combining the advantages of long-read and short-read technologies.


