
In condensed matter physics, a Bose–Einstein condensate (BEC) is a state of matter that is typically formed when a gas of bosons at very low densities is cooled to temperatures very close to absolute zero, i.e., 0 K. Under such conditions, a large fraction of bosons occupy the lowest quantum state, at which microscopic quantum-mechanical phenomena, particularly wavefunction interference, become apparent macroscopically. More generally, condensation refers to the appearance of macroscopic occupation of one or several states: for example, in BCS theory, a superconductor is a condensate of Cooper pairs. As such, condensation can be associated with phase transition, and the macroscopic occupation of the state is the order parameter.
Electronegativity, symbolized as χ, is the tendency for an atom of a given chemical element to attract shared electrons when forming a chemical bond. An atom's electronegativity is affected by both its atomic number and the distance at which its valence electrons reside from the charged nucleus. The higher the associated electronegativity, the more an atom or a substituent group attracts electrons. Electronegativity serves as a simple way to quantitatively estimate the bond energy, and the sign and magnitude of a bond's chemical polarity, which characterizes a bond along the continuous scale from covalent to ionic bonding. The loosely defined term electropositivity is the opposite of electronegativity: it characterizes an element's tendency to donate valence electrons.

In physics and chemistry, an equation of state is a thermodynamic equation relating state variables, which describe the state of matter under a given set of physical conditions, such as pressure, volume, temperature, or internal energy. Most modern equations of state are formulated in the Helmholtz free energy. Equations of state are useful in describing the properties of pure substances and mixtures in liquids, gases, and solid states as well as the state of matter in the interior of stars.
In thermodynamics, the specific heat capacity of a substance is the amount of heat that must be added to one unit of mass of the substance in order to cause an increase of one unit in temperature. It is also referred to as Massic heat capacity or as the Specific heat. More formally it is the heat capacity of a sample of the substance divided by the mass of the sample. The SI unit of specific heat capacity is joule per kelvin per kilogram, J⋅kg−1⋅K−1. For example, the heat required to raise the temperature of 1 kg of water by 1 K is 4184 joules, so the specific heat capacity of water is 4184 J⋅kg−1⋅K−1.

In thermodynamics, the enthalpy of vaporization, also known as the (latent) heat of vaporization or heat of evaporation, is the amount of energy (enthalpy) that must be added to a liquid substance to transform a quantity of that substance into a gas. The enthalpy of vaporization is a function of the pressure and temperature at which the transformation takes place.

Vapor pressure or equilibrium vapor pressure is the pressure exerted by a vapor in thermodynamic equilibrium with its condensed phases at a given temperature in a closed system. The equilibrium vapor pressure is an indication of a liquid's thermodynamic tendency to evaporate. It relates to the balance of particles escaping from the liquid in equilibrium with those in a coexisting vapor phase. A substance with a high vapor pressure at normal temperatures is often referred to as volatile. The pressure exhibited by vapor present above a liquid surface is known as vapor pressure. As the temperature of a liquid increases, the attractive interactions between liquid molecules become less significant in comparison to the entropy of those molecules in the gas phase, increasing the vapor pressure. Thus, liquids with strong intermolecular interactions are likely to have smaller vapor pressures, with the reverse true for weaker interactions.
The thermal conductivity of a material is a measure of its ability to conduct heat. It is commonly denoted by , , or and is measured in W·m−1·K−1.

In chemistry, the molar mass of a chemical compound is defined as the ratio between the mass and the amount of substance of any sample of the compound. The molar mass is a bulk, not molecular, property of a substance. The molar mass is an average of many instances of the compound, which often vary in mass due to the presence of isotopes. Most commonly, the molar mass is computed from the standard atomic weights and is thus a terrestrial average and a function of the relative abundance of the isotopes of the constituent atoms on Earth. The molar mass is appropriate for converting between the mass of a substance and the amount of a substance for bulk quantities.
Density functional theory (DFT) is a computational quantum mechanical modelling method used in physics, chemistry and materials science to investigate the electronic structure of many-body systems, in particular atoms, molecules, and the condensed phases. Using this theory, the properties of a many-electron system can be determined by using functionals - that is, functions that accept a function as input and output a single real number. In the case of DFT, these are functionals of the spatially dependent electron density. DFT is among the most popular and versatile methods available in condensed-matter physics, computational physics, and computational chemistry.
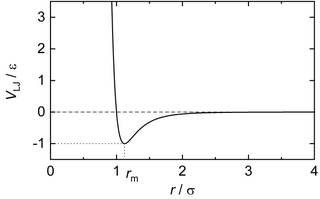
In computational chemistry, molecular physics, and physical chemistry, the Lennard-Jones potential is an intermolecular pair potential. Out of all the intermolecular potentials, the Lennard-Jones potential is probably the one that has been the most extensively studied. It is considered an archetype model for simple yet realistic intermolecular interactions. The Lennard-Jones potential is often used as a building block in molecular models for more complex substances. Many studies of the idealized "Lennard-Jones substance" use the potential to understand the physical nature of matter.
The Ising model, named after the physicists Ernst Ising and Wilhelm Lenz, is a mathematical model of ferromagnetism in statistical mechanics. The model consists of discrete variables that represent magnetic dipole moments of atomic "spins" that can be in one of two states. The spins are arranged in a graph, usually a lattice, allowing each spin to interact with its neighbors. Neighboring spins that agree have a lower energy than those that disagree; the system tends to the lowest energy but heat disturbs this tendency, thus creating the possibility of different structural phases. The model allows the identification of phase transitions as a simplified model of reality. The two-dimensional square-lattice Ising model is one of the simplest statistical models to show a phase transition.
The Born–Haber cycle is an approach to analyze reaction energies. It was named after two German scientists, Max Born and Fritz Haber, who developed it in 1919. It was also independently formulated by Kasimir Fajans and published concurrently in the same journal. The cycle is concerned with the formation of an ionic compound from the reaction of a metal with a halogen or other non-metallic element such as oxygen.
In thermodynamics, the heat transfer coefficient or film coefficient, or film effectiveness, is the proportionality constant between the heat flux and the thermodynamic driving force for the flow of heat. It is used in calculating the heat transfer, typically by convection or phase transition between a fluid and a solid. The heat transfer coefficient has SI units in watts per square meter per kelvin.
In polymer chemistry, the molar mass distribution describes the relationship between the number of moles of each polymer species and the molar mass of that species. In linear polymers, the individual polymer chains rarely have exactly the same degree of polymerization and molar mass, and there is always a distribution around an average value. The molar mass distribution of a polymer may be modified by polymer fractionation.
Static light scattering is a technique in physical chemistry that measures the intensity of the scattered light to obtain the average molecular weight Mw of a macromolecule like a polymer or a protein in solution. Measurement of the scattering intensity at many angles allows calculation of the root mean square radius, also called the radius of gyration Rg. By measuring the scattering intensity for many samples of various concentrations, the second virial coefficient, A2, can be calculated.
The Joback method, often named Joback–Reid method, predicts eleven important and commonly used pure component thermodynamic properties from molecular structure only. It is named after Kevin G. Joback in 1984 and developed it further with Robert C. Reid. The Joback method is an extension of the Lydersen method and uses very similar groups, formulas, and parameters for the three properties the Lydersen already supported.
A group-contribution method in chemistry is a technique to estimate and predict thermodynamic and other properties from molecular structures.

Viscosity is a measure of a fluid's rate-dependent resistance to a change in shape or to movement of its neighboring portions relative to one another. For liquids, it corresponds to the informal concept of thickness; for example, syrup has a higher viscosity than water. Viscosity is defined scientifically as a force multiplied by a time divided by an area. Thus its SI units are newton-seconds per square meter, or pascal-seconds.
The Lydersen method is a group contribution method for the estimation of critical properties temperature (Tc), pressure (Pc) and volume (Vc). The method is named after Aksel Lydersen who published it in 1955. The Lydersen method is the prototype for and ancestor of many new models like Joback, Klincewicz, Ambrose, Gani-Constantinou and others.
In polymer chemistry and polymer physics, the Flory–Fox equation is a simple empirical formula that relates molecular weight to the glass transition temperature of a polymer system. The equation was first proposed in 1950 by Paul J. Flory and Thomas G. Fox while at Cornell University. Their work on the subject overturned the previously held theory that the glass transition temperature was the temperature at which viscosity reached a maximum. Instead, they demonstrated that the glass transition temperature is the temperature at which the free space available for molecular motions achieved a minimum value. While its accuracy is usually limited to samples of narrow range molecular weight distributions, it serves as a good starting point for more complex structure-property relationships.










