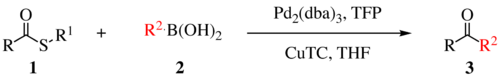The Liebeskind–Srogl coupling reaction is an organic reaction forming a new carbon–carbon bond from a thioester and a boronic acid using a metal catalyst. It is a cross-coupling reaction.[1] This reaction was invented by and named after Jiri Srogl from the Academy of Sciences, Czech Republic, and Lanny S. Liebeskind from Emory University, Atlanta, Georgia, USA. There are three generations of this reaction, with the first generation shown below. The original transformation used catalytic Pd(0), TFP = tris(2-furyl)phosphine as an additional ligand and stoichiometric CuTC = copper(I) thiophene-2-carboxylate as a co-metal catalyst. The overall reaction scheme is shown below.
The Liebeskind-Srogl coupling reaction
Liebeskind-Srogl reaction is most commonly seen with sulfide or thioester electrophiles and boronic acid or stannane nucleophiles but many other coupling partners are viable. In addition to alkyl and aryl thioesters; (hetero)aryl sulfides, thioamides, sulfanyl alkynes, and thiocyanates are competent electrophiles.[2] Virtually any metal-R bond capable of transmetalation has been demonstrated.[2] Indium derived nucleophiles require no copper or base. Note that this scope is applicable for the first generation coupling as the second and third generations are mechanistically distinct and have only been demonstrated with thioesters capable of forming the six-membered metallocycle, boronic acids, and stannanes.
The first-generation approach to cross coupling is run under anaerobic conditions using stoichiometric copper and catalytic palladium.[1]
Second generation approach renders the reactions catalytic in copper by using an extra equivalent of boronic acid under aerobic, palladium free conditions.[3] The additional equivalent liberates the copper from the sulfur auxiliary and allows it to turn over. This chemistry is limited to thioesters and sulfides and could also be limited by the cost and availability of the organoboron reagent.
The third generation renders the reaction catalytic in copper while using only one equivalent of boronic acid.[4]
Mechanism
Generation 1
The proposed reaction mechanism for the first generation is shown below.[5][6] The thioester 1 complexes with copper complex 3 to form compound 4. With the oxidative insertion of [Pd] into the carbon–sulfur bond, compound 5 is formed, and with transmetallation, organopalladium species 8 is formed. The transmetallation proceeds via the transfer of R2 to the palladium metal center with concomitant transfer of the sulfur atom to the copper complex. Reductive elimination gives ketone 3 with the regeneration of the active catalyst 9.
The Liebeskind–Srogl coupling mechanism
Generation 2
The mechanism for the second generation is shown below.[3] The mechanism does not follow a traditional oxidative addition-transmetelation-reductive elimination pathway like the first generation. In parallel to studies of Cu(I)-dioxygen reactions, a higher oxidation state, Cu-templated coupling is proposed.[7][8][9][10][11] Coordination of copper(I) to the thioester undergoes oxidation by air to give a copper (II/III) intermediate. Metal templating by Cu(II/III) acts as a Lewis acid to both activate the thiol ester and deliver R2 (from either boron directly or via an intermediate Cu-R2 species), which produces the ketone and a Cu-thiolate. A second equivalent of boronic acid is needed to break the copper sulfur bond and liberate copper back into the catalytic cycle.
Generation 3
The third generation renders the reaction catalytic in copper and uses only one equivalent of boronic acid by mimicking the metallothionein (MT) system that sponges metals from biological systems.[4] The thio-auxiliary features an N–O motif that mimics the S–S motif in the MT biosystem, that is necessary to break the copper sulfur bond and turn over the catalyst. This generation is palladium free and under microwave conditions. The mechanism is expected to follow that of the second generation (shown as an active Cu(I)-R2 species but R2 could be delivered directly from the coordinated boronic acid) but includes the auxiliary releasing copper back into the catalytic cycle instead of additional boronic acid.
Applications in synthesis
The Liebeskind–Srogl coupling has been used as a key retrosynthetic disconnection in several natural product total synthesis.
For example, in the synthesis of Goniodomin A, the Sasaki lab utilized this chemistry to rapidly access the northern half of the natural product.[12]
The Guerrero lab used the Liebeskind–Srogl coupling to construct the entire carbon skeleton of viridin in high yield on multi-gram scale.[13]
The lab of Figadere used the Liebeskind–Srogl coupling early in their synthesis of amphidinolide F[14] by employing this reaction to construct the north eastern fragment of the macrocycle and the terpene chain.
Other
Directed difunctionalization
The Yu lab has demonstrated that in the presence of two sulfide bonds, one can be selectively functionalized in the presence of one equivalent of nucleophile if directed by a carbonyl oxygen.[15] This reaction proceeds through a five-membered palladacycle with oxidative addition taking place on this cis-thioether. Additional equivalence of nucleophile will functionalize the trans-position.
↑ Yu, Y.; Liebeskind, L. S. (2004). "Copper-mediated, palladium-catalyzed coupling of thiol esters with aliphatic organoboron reagents". J. Org. Chem.69 (10): 3554–3557. doi:10.1021/jo049964p. PMID15132570.
↑ Hatcher, Lanying Q.; Vance, Michael A.; Narducci Sarjeant, Amy A.; Solomon, Edward I.; Karlin, Kenneth D. (April 2006). "Copper−Dioxygen Adducts and the Side-on Peroxo Dicopper(II)/Bis(μ-oxo) Dicopper(III) Equilibrium: Significant Ligand Electronic Effects". Inorganic Chemistry. 45 (7): 3004–3013. doi:10.1021/ic052185m. ISSN0020-1669. PMID16562956.
↑ Mirica, Liviu M.; Rudd, Deanne Jackson; Vance, Michael A.; Solomon, Edward I.; Hodgson, Keith O.; Hedman, Britt; Stack, T. Daniel P. (March 2006). "μ-η2:η2-Peroxodicopper(II) Complex with a Secondary Diamine Ligand: A Functional Model of Tyrosinase". Journal of the American Chemical Society. 128 (8): 2654–2665. Bibcode:2006JAChS.128.2654M. doi:10.1021/ja056740v. ISSN0002-7863. PMID16492052.
↑ Matsumoto, Takahiro; Furutachi, Hideki; Kobino, Masashi; Tomii, Masato; Nagatomo, Shigenori; Tosha, Takehiko; Osako, Takao; Fujinami, Shuhei; Itoh, Shinobu (March 2006). "Intramolecular Arene Hydroxylation versus Intermolecular Olefin Epoxidation by (μ-η2:η2-Peroxo)dicopper(II) Complex Supported by Dinucleating Ligand". Journal of the American Chemical Society. 128 (12): 3874–3875. Bibcode:2006JAChS.128.3874M. doi:10.1021/ja058117g. ISSN0002-7863. PMID16551071.
↑ Lewis, Elizabeth A.; Tolman, William B. (February 2004). "Reactivity of Dioxygen−Copper Systems". Chemical Reviews. 104 (2): 1047–1076. doi:10.1021/cr020633r. ISSN0009-2665. PMID14871149.
↑ Saito, Tomoyuki; Fuwa, Haruhiko; Sasaki, Makoto (19 November 2009). "Toward the Total Synthesis of Goniodomin A, An Actin-Targeting Marine Polyether Macrolide: Convergent Synthesis of the C15−C36 Segment". Organic Letters. 11 (22): 5274–5277. doi:10.1021/ol902217q. ISSN1523-7060. PMID19905029.
↑ Del Bel, Matthew; Abela, Alexander R.; Ng, Jeffrey D.; Guerrero, Carlos A. (24 May 2017). "Enantioselective Chemical Syntheses of the Furanosteroids (−)-Viridin and (−)-Viridiol". Journal of the American Chemical Society. 139 (20): 6819–6822. Bibcode:2017JAChS.139.6819D. doi:10.1021/jacs.7b02829. ISSN0002-7863. PMID28463562.
↑ Jin, Weiwei; Du, Wangming; Yang, Qin; Yu, Haifeng; Chen, Jiping; Yu, Zhengkun (19 August 2011). "Regio- and Stereoselective Synthesis of Multisubstituted Olefins and Conjugate Dienes by Using α-Oxo Ketene Dithioacetals as the Building Blocks". Organic Letters. 13 (16): 4272–4275. doi:10.1021/ol201620g. ISSN1523-7060. PMID21761823.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.