Related Research Articles

Brain abscess is an abscess within the brain tissue caused by inflammation and collection of infected material coming from local or remote infectious sources. The infection may also be introduced through a skull fracture following a head trauma or surgical procedures. Brain abscess is usually associated with congenital heart disease in young children. It may occur at any age but is most frequent in the third decade of life.

Progressive multifocal leukoencephalopathy (PML) is a rare and often fatal viral disease characterized by progressive damage (-pathy) or inflammation of the white matter (leuko-) of the brain (-encephalo-) at multiple locations (multifocal). It is caused by the JC virus, which is normally present and kept under control by the immune system. The JC virus is harmless except in cases of weakened immune systems. In general, PML has a mortality rate of 30–50% in the first few months, and those who survive can be left with varying degrees of neurological disabilities.
Porencephaly is an extremely rare cephalic disorder involving encephalomalacia. It is a neurological disorder of the central nervous system characterized by cysts or cavities within the cerebral hemisphere. Porencephaly was termed by Heschl in 1859 to describe a cavity in the human brain. Derived from Greek roots, the word porencephaly means 'holes in the brain'. The cysts and cavities are more likely to be the result of destructive (encephaloclastic) cause, but can also be from abnormal development (malformative), direct damage, inflammation, or hemorrhage. The cysts and cavities cause a wide range of physiological, physical, and neurological symptoms. Depending on the patient, this disorder may cause only minor neurological problems, without any disruption of intelligence, while others may be severely disabled or die before the second decade of their lives. However, this disorder is far more common within infants, and porencephaly can occur both before or after birth.

Tuberous sclerosis complex (TSC) is a rare multisystem autosomal dominant genetic disease that causes non-cancerous tumours to grow in the brain and on other vital organs such as the kidneys, heart, liver, eyes, lungs and skin. A combination of symptoms may include seizures, intellectual disability, developmental delay, behavioral problems, skin abnormalities, lung disease, and kidney disease.
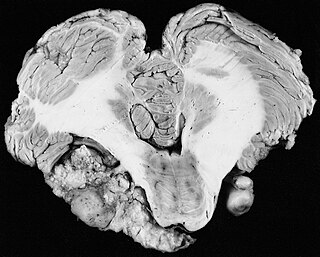
A vestibular schwannoma (VS), also called acoustic neuroma, is a benign tumor that develops on the vestibulocochlear nerve that passes from the inner ear to the brain. The tumor originates when Schwann cells that form the insulating myelin sheath on the nerve malfunction. Normally, Schwann cells function beneficially to protect the nerves which transmit balance and sound information to the brain. However, sometimes a mutation in the tumor suppressor gene, NF2, located on chromosome 22, results in abnormal production of the cell protein named Merlin, and Schwann cells multiply to form a tumor. The tumor originates mostly on the vestibular division of the nerve rather than the cochlear division, but hearing as well as balance will be affected as the tumor enlarges.

Neurofibromatosis type I (NF-1), or von Recklinghausen syndrome, is a complex multi-system human disorder caused by the mutation of Neurofibromin 1, a gene on chromosome 17 that is responsible for production of a protein (neurofibromin) which is needed for normal function in many human cell types. NF-1 causes tumors along the nervous system which can grow anywhere on the body. NF-1 is one of the most common genetic disorders and is not limited to any person's race or sex. NF-1 is an autosomal dominant disorder, which means that mutation or deletion of one copy of the NF-1 gene is sufficient for the development of NF-1, although presentation varies widely and is often different even between relatives affected by NF-1.

Neurofibromatosis type II is a genetic condition that may be inherited or may arise spontaneously, and causes benign tumors of the brain, spinal cord, and peripheral nerves. The types of tumors frequently associated with NF2 include vestibular schwannomas, meningiomas, and ependymomas. The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII, which is the "auditory-vestibular nerve" that transmits sensory information from the inner ear to the brain. Besides, other benign brain and spinal tumors occur. Symptoms depend on the presence, localisation and growth of the tumor(s), in which multiple cranial nerves can be involved. Many people with this condition also experience vision problems. Neurofibromatosis type II is caused by mutations of the "Merlin" gene, which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.
Phakomatoses, also known neurocutaneous syndromes, are a group of multisystemic diseases that most prominently affect structures primarily derived from the ectoderm such as the central nervous system, skin and eyes. The majority of phakomatoses are single-gene disorders that may be inherited in an autosomal dominant, autosomal recessive or X-linked pattern. Presentations may vary dramatically between patients with the same particular syndrome due to mosaicism, variable expressivity, and penetrance.
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an acquired autoimmune disease of the peripheral nervous system characterized by progressive weakness and impaired sensory function in the legs and arms. The disorder is sometimes called chronic relapsing polyneuropathy (CRP) or chronic inflammatory demyelinating polyradiculoneuropathy. CIDP is closely related to Guillain–Barré syndrome and it is considered the chronic counterpart of that acute disease. Its symptoms are also similar to progressive inflammatory neuropathy. It is one of several types of neuropathy.
Pseudobulbar palsy is a medical condition characterized by the inability to control facial movements and caused by a variety of neurological disorders. Patients experience difficulty chewing and swallowing, have increased reflexes and spasticity in tongue and the bulbar region, and demonstrate slurred speech, sometimes also demonstrating uncontrolled emotional outbursts.
Inflammatory demyelinating diseases (IDDs), sometimes called Idiopathic (IIDDs) due to the unknown etiology of some of them, are a heterogenous group of demyelinating diseases - conditions that cause damage to myelin, the protective sheath of nerve fibers - that occur against the background of an acute or chronic inflammatory process. IDDs share characteristics with and are often grouped together under Multiple Sclerosis. They are sometimes considered different diseases from Multiple Sclerosis, but considered by others to form a spectrum differing only in terms of chronicity, severity, and clinical course.

Limbic encephalitis is a form of encephalitis, a disease characterized by inflammation of the brain. Limbic encephalitis is caused by autoimmunity: an abnormal state where the body produces antibodies against itself. Some cases are associated with cancer and some are not. Although the disease is known as "limbic" encephalitis, it is seldom limited to the limbic system and post-mortem studies usually show involvement of other parts of the brain. The disease was first described by Brierley and others in 1960 as a series of three cases. The link to cancer was first noted in 1968 and confirmed by later investigators.

Multiple sclerosis can cause a variety of symptoms: changes in sensation (hypoesthesia), muscle weakness, abnormal muscle spasms, or difficulty moving; difficulties with coordination and balance; problems in speech (dysarthria) or swallowing (dysphagia), visual problems, fatigue and acute or chronic pain syndromes, bladder and bowel difficulties, cognitive impairment, or emotional symptomatology. The main clinical measure in progression of the disability and severity of the symptoms is the Expanded Disability Status Scale or EDSS.

Tumefactive multiple sclerosis is a condition in which the central nervous system of a person has multiple demyelinating lesions with atypical characteristics for those of standard multiple sclerosis (MS). It is called tumefactive as the lesions are "tumor-like" and they mimic tumors clinically, radiologically and sometimes pathologically.

Bonnet–Dechaume–Blanc syndrome, also known as Wyburn-Mason syndrome, is a rare congenital disorder characterized by arteriovenous malformations of the brain, retina or facial nevi. The syndrome has a number of possible symptoms and can, more rarely, affect the skin, bones, kidneys, muscles, and gastrointestinal tract. When the syndrome affects the brain, people can experience severe headaches, seizures, acute stroke, meningism, and progressive neurological deficits due to acute or chronic ischaemia caused by arteriovenous shunting.
Behçet's disease is recognized as a disease that cause inflammatory perivasculitis, inflammation of the tissue around a blood or lymph vessel, in practically any tissue in the body. Usually, prevalent symptoms include canker sores or ulcers in the mouth and on the genitals, and inflammation in parts of the eye. In addition, patients experience severe headache and papulopustular skin lesions as well. The disease was first described in 1937 by a Turkish dermatologist, Dr. Hulusi Behçet. Behçet's disease is most prevalent in the Middle East and the Far East regions; however, it is rare in America regions.

A brain metastasis is a cancer that has metastasized (spread) to the brain from another location in the body and is therefore considered a secondary brain tumor. The metastasis typically shares a cancer cell type with the original site of the cancer. Metastasis is the most common cause of brain cancer, as primary tumors that originate in the brain are less common. The most common sites of primary cancer which metastasize to the brain are lung, breast, colon, kidney, and skin cancer. Brain metastases can occur in patients months or even years after their original cancer is treated. Brain metastases have a poor prognosis for cure, but modern treatments are allowing patients to live months and sometimes years after the diagnosis.

Cavernous hemangioma, also called cavernous angioma, venous malformation, or cavernoma, is a type of venous malformation due to endothelial dysmorphogenesis from a lesion which is present at birth. A cavernoma in the brain is called a cerebral cavernous malformation or CCM. Despite its designation as a hemangioma, a cavernous hemangioma is not a tumor as it does not display endothelial hyperplasia. The abnormal tissue causes a slowing of blood flow through the cavities, or "caverns". The blood vessels do not form the necessary junctions with surrounding cells, and the structural support from the smooth muscle is hindered, causing leakage into the surrounding tissue. It is the leakage of blood, referred to as hemorrhage, that causes a variety of symptoms known to be associated with the condition.
MOG antibody disease (MOGAD) or MOG antibody-associated encephalomyelitis (MOG-EM) is an inflammatory demyelinating disease of the central nervous system. Serum anti-myelin oligodendrocyte glycoprotein antibodies are present in up to half of patients with an acquired demyelinating syndrome and have been described in association with a range of phenotypic presentations, including acute disseminated encephalomyelitis, optic neuritis, transverse myelitis, and neuromyelitis optica.

Angiocentric glioma (AG) refers to a rare neuroepithelial tumor when the superficial brain malignant cells enclose the brain vessels, commonly found in children and young adults. Initially identified in 2005 by Wang and his team from the University of Texas, AG was classified as Grade I by 2007 WHO Classification of Tumors of the Central Nervous System due to its benign clinical behavior, low proliferation index, and curative properties. AG primarily affects children and young adults at an average initial diagnosis age of 16 years old. Over 85% AG patients experience intractable seizures since childhood, especially partial epilepsy.
References
- ↑ Wiebe, S. (1 April 1999). "Meningioangiomatosis: A comprehensive analysis of clinical and laboratory features". Brain. 122 (4): 709–726. doi: 10.1093/brain/122.4.709 . PMID 10219783.
- ↑ Jamil O, Ramkissoon S, Folkerth R, Smith E (2012). "Multifocal meningioangiomatosis in a 3-year-old patient". Journal of Neurosurgery: Pediatrics (Case report). 10 (6): 486–9. doi:10.3171/2012.9.PEDS1224. PMC 3762590 . PMID 23020197.