Related Research Articles

Glycogen storage disease type V, also known as McArdle's disease, is a metabolic disorder, one of the metabolic myopathies, more specifically a muscle glycogen storage disease, caused by a deficiency of myophosphorylase. Its incidence is reported as one in 100,000, roughly the same as glycogen storage disease type I.

Glucagon is a peptide hormone, produced by alpha cells of the pancreas. It raises the concentration of glucose and fatty acids in the bloodstream and is considered to be the main catabolic hormone of the body. It is also used as a medication to treat a number of health conditions. Its effect is opposite to that of insulin, which lowers extracellular glucose. It is produced from proglucagon, encoded by the GCG gene.
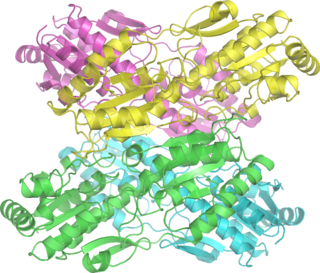
Phosphofructokinase-1 (PFK-1) is one of the most important regulatory enzymes of glycolysis. It is an allosteric enzyme made of 4 subunits and controlled by many activators and inhibitors. PFK-1 catalyzes the important "committed" step of glycolysis, the conversion of fructose 6-phosphate and ATP to fructose 1,6-bisphosphate and ADP. Glycolysis is the foundation for respiration, both anaerobic and aerobic. Because phosphofructokinase (PFK) catalyzes the ATP-dependent phosphorylation to convert fructose-6-phosphate into fructose 1,6-bisphosphate and ADP, it is one of the key regulatory steps of glycolysis. PFK is able to regulate glycolysis through allosteric inhibition, and in this way, the cell can increase or decrease the rate of glycolysis in response to the cell's energy requirements. For example, a high ratio of ATP to ADP will inhibit PFK and glycolysis. The key difference between the regulation of PFK in eukaryotes and prokaryotes is that in eukaryotes PFK is activated by fructose 2,6-bisphosphate. The purpose of fructose 2,6-bisphosphate is to supersede ATP inhibition, thus allowing eukaryotes to have greater sensitivity to regulation by hormones like glucagon and insulin.

Pyruvate kinase was inappropriately named before it was recognized that it did not directly catalyze phosphorylation of pyruvate, which does not occur under physiological conditions. Pyruvate kinase is present in four distinct, tissue-specific isozymes in animals, each consisting of particular kinetic properties necessary to accommodate the variations in metabolic requirements of diverse tissues.
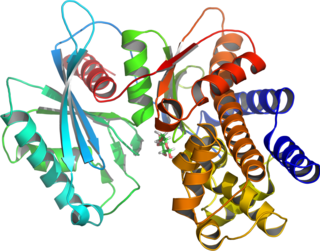
Glucokinase is an enzyme that facilitates phosphorylation of glucose to glucose-6-phosphate. Glucokinase occurs in cells in the liver and pancreas of humans and most other vertebrates. In each of these organs it plays an important role in the regulation of carbohydrate metabolism by acting as a glucose sensor, triggering shifts in metabolism or cell function in response to rising or falling levels of glucose, such as occur after a meal or when fasting. Mutations of the gene for this enzyme can cause unusual forms of diabetes or hypoglycemia.

In biochemistry, phosphorylases are enzymes that catalyze the addition of a phosphate group from an inorganic phosphate (phosphate+hydrogen) to an acceptor.
Glycogenesis is the process of glycogen synthesis, in which glucose molecules are added to chains of glycogen for storage. This process is activated during rest periods following the Cori cycle, in the liver, and also activated by insulin in response to high glucose levels.

Glycogen phosphorylase is one of the phosphorylase enzymes. Glycogen phosphorylase catalyzes the rate-limiting step in glycogenolysis in animals by releasing glucose-1-phosphate from the terminal alpha-1,4-glycosidic bond. Glycogen phosphorylase is also studied as a model protein regulated by both reversible phosphorylation and allosteric effects.
Glycogen storage disease type I is an inherited disease that prevents the liver from properly breaking down stored glycogen, which is necessary to maintain adequate blood sugar levels. GSD I is divided into two main types, GSD Ia and GSD Ib, which differ in cause, presentation, and treatment. There are also possibly rarer subtypes, the translocases for inorganic phosphate or glucose ; however, a recent study suggests that the biochemical assays used to differentiate GSD Ic and GSD Id from GSD Ib are not reliable, and are therefore GSD Ib.
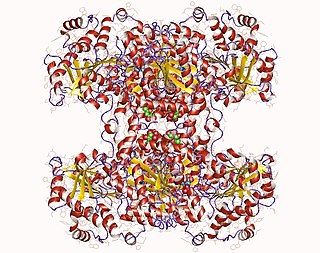
Glycogen synthase is a key enzyme in glycogenesis, the conversion of glucose into glycogen. It is a glycosyltransferase that catalyses the reaction of UDP-glucose and n to yield UDP and n+1.

The glycogen debranching enzyme, in humans, is the protein encoded by the gene AGL. This enzyme is essential for the breakdown of glycogen, which serves as a store of glucose in the body. It has separate glucosyltransferase and glucosidase activities.

The enzyme glucose 6-phosphatase (EC 3.1.3.9, G6Pase; systematic name D-glucose-6-phosphate phosphohydrolase) catalyzes the hydrolysis of glucose 6-phosphate, resulting in the creation of a phosphate group and free glucose:

1,4-alpha-glucan-branching enzyme, also known as brancher enzyme or glycogen-branching enzyme is an enzyme that in humans is encoded by the GBE1 gene.

Myophosphorylase or glycogen phosphorylase, muscle associated (PYGM) is the muscle isoform of the enzyme glycogen phosphorylase and is encoded by the PYGM gene. This enzyme helps break down glycogen into glucose-1-phosphate, so it can be used within the muscle cell. Mutations in this gene are associated with McArdle disease, a glycogen storage disease of muscle.
Enzyme activators are molecules that bind to enzymes and increase their activity. They are the opposite of enzyme inhibitors. These molecules are often involved in the allosteric regulation of enzymes in the control of metabolism. An example of an enzyme activator working in this way is fructose 2,6-bisphosphate, which activates phosphofructokinase 1 and increases the rate of glycolysis in response to the hormone glucagon. In some cases, when a substrate binds to one catalytic subunit of an enzyme, this can trigger an increase in the substrate affinity as well as catalytic activity in the enzyme's other subunits, and thus the substrate acts as an activator.

Glucose-6-phosphate exchanger SLC37A4, also known as glucose-6-phosphate translocase, is an enzyme that in humans is encoded by the SLC37A4 gene.

6-phosphofructokinase, muscle type is an enzyme that in humans is encoded by the PFKM gene on chromosome 12. Three phosphofructokinase isozymes exist in humans: muscle, liver and platelet. These isozymes function as subunits of the mammalian tetramer phosphofructokinase, which catalyzes the phosphorylation of fructose-6-phosphate to fructose-1,6-bisphosphate. Tetramer composition varies depending on tissue type. This gene encodes the muscle-type isozyme. Mutations in this gene have been associated with glycogen storage disease type VII, also known as Tarui disease. Alternatively spliced transcript variants have been described.[provided by RefSeq, Nov 2009]

Phosphorylase b kinase gamma catalytic chain, testis/liver isoform is an enzyme that in humans is encoded by the PHKG2 gene.

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.
Glycogen phosphorylase, brain, is an enzyme that in humans is encoded by the PYGB gene on chromosome 20. The protein encoded by this gene is a glycogen phosphorylase found predominantly in the brain. The encoded protein forms homodimers which can associate into homotetramers, the enzymatically active form of glycogen phosphorylase. The activity of this enzyme is positively regulated by AMP and negatively regulated by ATP, ADP, and glucose-6-phosphate. This enzyme catalyzes the rate-determining step in glycogen degradation. [provided by RefSeq, Jul 2008]
References
- 1 2 "PYGL phosphorylase, glycogen, liver". NCBI Entrez Gene database.
- ↑ "UniProtKB: P06737".
- 1 2 3 Newgard, CB; Littman, DR; van Genderen, C; Smith, M; Fletterick, RJ (15 March 1988). "Human brain glycogen phosphorylase. Cloning, sequence analysis, chromosomal mapping, tissue expression, and comparison with the human liver and muscle isozymes". The Journal of Biological Chemistry. 263 (8): 3850–7. doi: 10.1016/S0021-9258(18)69003-9 . PMID 3346228.
- 1 2 3 4 5 Chang, S; Rosenberg, MJ; Morton, H; Francomano, CA; Biesecker, LG (May 1998). "Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI". Human Molecular Genetics. 7 (5): 865–70. doi: 10.1093/hmg/7.5.865 . PMID 9536091.
- 1 2 3 4 5 6 7 8 9 10 Rath, VL; Ammirati, M; LeMotte, PK; Fennell, KF; Mansour, MN; Danley, DE; Hynes, TR; Schulte, GK; Wasilko, DJ; Pandit, J (July 2000). "Activation of human liver glycogen phosphorylase by alteration of the secondary structure and packing of the catalytic core". Molecular Cell. 6 (1): 139–48. doi: 10.1016/s1097-2765(00)00015-0 . PMID 10949035.
- 1 2 3 4 Ekstrom, JL; Pauly, TA; Carty, MD; Soeller, WC; Culp, J; Danley, DE; Hoover, DJ; Treadway, JL; Gibbs, EM; Fletterick, RJ; Day, YS; Myszka, DG; Rath, VL (August 2002). "Structure-activity analysis of the purine binding site of human liver glycogen phosphorylase". Chemistry & Biology. 9 (8): 915–24. doi: 10.1016/s1074-5521(02)00186-2 . PMID 12204691.
- 1 2 3 4 5 6 Burwinkel, B; Bakker, HD; Herschkovitz, E; Moses, SW; Shin, YS; Kilimann, MW (April 1998). "Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI". American Journal of Human Genetics. 62 (4): 785–91. doi:10.1086/301790. PMC 1377030 . PMID 9529348.
- ↑ Gall, D; Baus, E; Dupont, G (21 December 2000). "Activation of the liver glycogen phosphorylase by Ca(2+)oscillations: a theoretical study". Journal of Theoretical Biology. 207 (4): 445–54. Bibcode:2000JThBi.207..445G. doi:10.1006/jtbi.2000.2139. PMID 11093832.
- ↑ Newgard, CB; Nakano, K; Hwang, PK; Fletterick, RJ (November 1986). "Sequence analysis of the cDNA encoding human liver glycogen phosphorylase reveals tissue-specific codon usage". Proceedings of the National Academy of Sciences of the United States of America. 83 (21): 8132–6. Bibcode:1986PNAS...83.8132N. doi: 10.1073/pnas.83.21.8132 . PMC 386881 . PMID 2877458.
- ↑ Gelinas, RP; Froman, BE; McElroy, F; Tait, RC; Gorin, FA (November 1989). "Human brain glycogen phosphorylase: characterization of fetal cDNA and genomic sequences". Brain Research. Molecular Brain Research. 6 (2–3): 177–85. doi:10.1016/0169-328x(89)90052-1. PMID 2615594.
- 1 2 3 Tomihira, M; Kawasaki, E; Nakajima, H; Imamura, Y; Sato, Y; Sata, M; Kage, M; Sugie, H; Nunoi, K (August 2004). "Intermittent and recurrent hepatomegaly due to glycogen storage in a patient with type 1 diabetes: genetic analysis of the liver glycogen phosphorylase gene (PYGL)". Diabetes Research and Clinical Practice. 65 (2): 175–82. doi:10.1016/j.diabres.2003.12.004. PMID 15223230.