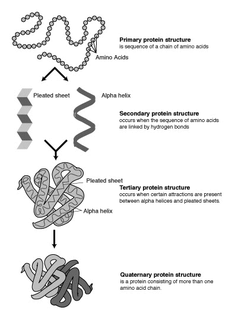
The alpha helix (α-helix) is a common motif in the secondary structure of proteins and is a right hand-helix conformation in which every backbone N−H group donates a hydrogen bond to the backbone C=O group of the amino acid located three or four residues earlier along the protein sequence.

The β-sheet is a common motif of regular secondary structure in proteins. Beta sheets consist of beta strands connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in formation of the protein aggregates and fibrils observed in many human diseases, notably the amyloidoses such as Alzheimer's disease.

Protein secondary structure is the three dimensional form of local segments of proteins. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its folding and its secondary and tertiary structure from its primary structure. Structure prediction is fundamentally different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by bioinformatics and theoretical chemistry; it is highly important in medicine and biotechnology. Every two years, the performance of current methods is assessed in the CASP experiment. A continuous evaluation of protein structure prediction web servers is performed by the community project CAMEO3D.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

A Ramachandran plot, originally developed in 1963 by G. N. Ramachandran, C. Ramakrishnan, and V. Sasisekharan, is a way to visualize energetically allowed regions for backbone dihedral angles ψ against φ of amino acid residues in protein structure. The figure at left illustrates the definition of the φ and ψ backbone dihedral angles. The ω angle at the peptide bond is normally 180°, since the partial-double-bond character keeps the peptide planar. The figure at top right shows the allowed φ,ψ backbone conformational regions from the Ramachandran et al. 1963 and 1968 hard-sphere calculations: full radius in solid outline, reduced radius in dashed, and relaxed tau (N-Cα-C) angle in dotted lines. Because dihedral angle values are circular and 0° is the same as 360°, the edges of the Ramachandran plot "wrap" right-to-left and bottom-to-top. For instance, the small strip of allowed values along the lower-left edge of the plot are a continuation of the large, extended-chain region at upper left.
Nuclear magnetic resonance spectroscopy of proteins is a field of structural biology in which NMR spectroscopy is used to obtain information about the structure and dynamics of proteins, and also nucleic acids, and their complexes. The field was pioneered by Richard R. Ernst and Kurt Wüthrich at the ETH, and by Ad Bax, Marius Clore, and Angela Gronenborn at the NIH, among others. Structure determination by NMR spectroscopy usually consists of several phases, each using a separate set of highly specialized techniques. The sample is prepared, measurements are made, interpretive approaches are applied, and a structure is calculated and validated.
ICM stands for Internal Coordinate Mechanics and was first designed and built to predict low-energy conformations of molecules by sampling the space of internal coordinates defining molecular geometry. In ICM each molecule is constructed as a tree from an entry atom where each next atom is built iteratively from the preceding three atoms via three internal variables. The rings kept rigid or imposed via additional restraints.
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.
A polyproline helix is a type of protein secondary structure which occurs in proteins comprising repeating proline residues. A left-handed polyproline II helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have trans isomers of their peptide bonds. This PPII conformation is also common in proteins and polypeptides with other amino acids apart from proline. Similarly, a more compact right-handed polyproline I helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have cis isomers of their peptide bonds. Of the twenty common naturally occurring amino acids, only proline is likely to adopt the cis isomer of the peptide bond, specifically the X-Pro peptide bond; steric and electronic factors heavily favor the trans isomer in most other peptide bonds. However, peptide bonds that replace proline with another N-substituted amino acid are also likely to adopt the cis isomer.
Conformational entropy is the entropy associated with the number of conformations of a molecule. The concept is most commonly applied to biological macromolecules such as proteins and RNA, but also be used for polysaccharides and other molecules. To calculate the conformational entropy, the possible conformations of the molecule may first be discretized into a finite number of states, usually characterized by unique combinations of certain structural parameters, each of which has been assigned an energy. In proteins, backbone dihedral angles and side chain rotamers are commonly used as parameters, and in RNA the base pairing pattern may be used. These characteristics are used to define the degrees of freedom. The conformational entropy associated with a particular structure or state, such as an alpha-helix, a folded or an unfolded protein structure, is then dependent on the probability of the occupancy of that structure.
Loop modeling is a problem in protein structure prediction requiring the prediction of the conformations of loop regions in proteins with or without the use of a structural template. Computer programs that solve these problems have been used to research a broad range of scientific topics from ADP to breast cancer. Because protein function is determined by its shape and the physiochemical properties of its exposed surface, it is important to create an accurate model for protein/ligand interaction studies. The problem arises often in homology modeling, where the tertiary structure of an amino acid sequence is predicted based on a sequence alignment to a template, or a second sequence whose structure is known. Because loops have highly variable sequences even within a given structural motif or protein fold, they often correspond to unaligned regions in sequence alignments; they also tend to be located at the solvent-exposed surface of globular proteins and thus are more conformationally flexible. Consequently, they often cannot be modeled using standard homology modeling techniques. More constrained versions of loop modeling are also used in the data fitting stages of solving a protein structure by X-ray crystallography, because loops can correspond to regions of low electron density and are therefore difficult to resolve.
FoldX is a protein design algorithm that uses an empirical force field. It can determine the energetic effect of point mutations as well as the interaction energy of protein complexes. FoldX can mutate protein and DNA side chains using a probability-based rotamer library, while exploring alternative conformations of the surrounding side chains.
I-sites are short sequence-structure motifs that are mined from the Protein Data Bank (PDB) that correlate strongly with three-dimensional structural elements. These sequence-structure motifs are used for the local structure prediction of proteins. Local structure can be expressed as fragments or as backbone angles. Locations in the protein sequence that have high confidence I-sites predictions may be the initiation sites of folding. I-sites have also been identified as discrete models for folding pathways. I-sites consist of about 250 motifs. Each motif has an amino acid profile, a fragment structure and optionally, a 4-dimensional tensor of pairwise sequence covariance.
Graphical models have become powerful frameworks for protein structure prediction, protein–protein interaction, and free energy calculations for protein structures. Using a graphical model to represent the protein structure allows the solution of many problems including secondary structure prediction, protein-protein interactions, protein-drug interaction, and free energy calculations.
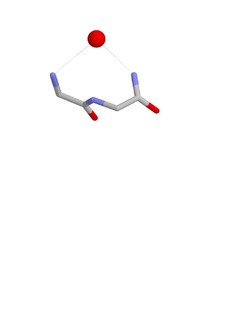
The Nest is a type of protein structural motif. It is a small recurring anion-binding feature of both proteins and peptides. Each consists of the main chain atoms of three consecutive amino acid residues. The main chain NH groups bind the anions while the side chain atoms are often not involved. Proline residues lack NH groups so are rare in nests. About one in 12 of amino acid residues in proteins, on average, belongs to a nest.

GeNMR method is the first fully automated template-based method of protein structure determination that utilizes both NMR chemical shifts and NOE -based distance restraints.
Volume, Area, Dihedral Angle Reporter (VADAR) is a freely available protein structure validation web server that was developed as a collaboration between Dr. Brian Sykes and Dr. David Wishart at the University of Alberta. VADAR consists of >15 different algorithms and programs for assessing and validating peptide and protein structures from their PDB coordinate data. VADAR is capable of determining secondary structure, identifying and classifying six different types of beta turns, determining and calculating the strength of C=O -- N-H hydrogen bonds, calculating residue-specific accessible surface areas (ASA), calculating residue volumes, determining backbone and side chain torsion angles, assessing local structure quality, evaluating global structure quality and identifying residue “outliers”. The results have been validated through extensive comparison to published data and careful visual inspection. VADAR produces both text and graphical output with most of the quantitative data presented in easily viewed tables. In particular, VADAR’s output is presented in a vertical, tabular format with most of the sequence data, residue numbering and any other calculated property or feature presented from top to bottom, rather than from left to right.