Related Research Articles
The Protein Data Bank (PDB) is a database for the three-dimensional structural data of large biological molecules such as proteins and nucleic acids, which is overseen by the Worldwide Protein Data Bank (wwPDB). These structural data are obtained and deposited by biologists and biochemists worldwide through the use of experimental methodologies such as X-ray crystallography, NMR spectroscopy, and, increasingly, cryo-electron microscopy. All submitted data are reviewed by expert biocurators and, once approved, are made freely available on the Internet under the CC0 Public Domain Dedication. Global access to the data is provided by the websites of the wwPDB member organisations.
The National Center for Biotechnology Information (NCBI) is part of the United States National Library of Medicine (NLM), a branch of the National Institutes of Health (NIH). It is approved and funded by the government of the United States. The NCBI is located in Bethesda, Maryland, and was founded in 1988 through legislation sponsored by US Congressman Claude Pepper.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
The Structural Classification of Proteins (SCOP) database is a largely manual classification of protein structural domains based on similarities of their structures and amino acid sequences. A motivation for this classification is to determine the evolutionary relationship between proteins. Proteins with the same shapes but having little sequence or functional similarity are placed in different superfamilies, and are assumed to have only a very distant common ancestor. Proteins having the same shape and some similarity of sequence and/or function are placed in "families", and are assumed to have a closer common ancestor.
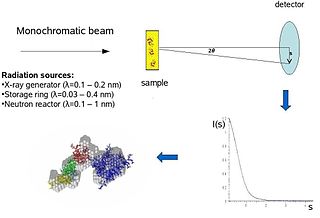
Biological small-angle scattering is a small-angle scattering method for structure analysis of biological materials. Small-angle scattering is used to study the structure of a variety of objects such as solutions of biological macromolecules, nanocomposites, alloys, and synthetic polymers. Small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS) are the two complementary techniques known jointly as small-angle scattering (SAS). SAS is an analogous method to X-ray and neutron diffraction, wide angle X-ray scattering, as well as to static light scattering. In contrast to other X-ray and neutron scattering methods, SAS yields information on the sizes and shapes of both crystalline and non-crystalline particles. When used to study biological materials, which are very often in aqueous solution, the scattering pattern is orientation averaged.

The CATH Protein Structure Classification database is a free, publicly available online resource that provides information on the evolutionary relationships of protein domains. It was created in the mid-1990s by Professor Christine Orengo and colleagues including Janet Thornton and David Jones, and continues to be developed by the Orengo group at University College London. CATH shares many broad features with the SCOP resource, however there are also many areas in which the detailed classification differs greatly.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, which are the monomers of the polymer. A single amino acid monomer may also be called a residue, which indicates a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions, such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo-electron microscopy (cryo-EM) and dual polarisation interferometry, to determine the structure of proteins.
A chemical file format is a type of data file which is used specifically for depicting molecular data. One of the most widely used is the chemical table file format, which is similar to Structure Data Format (SDF) files. They are text files that represent multiple chemical structure records and associated data fields. The XYZ file format is a simple format that usually gives the number of atoms in the first line, a comment on the second, followed by a number of lines with atomic symbols and cartesian coordinates. The Protein Data Bank Format is commonly used for proteins but is also used for other types of molecules. There are many other types which are detailed below. Various software systems are available to convert from one format to another.
RasMol is a computer program written for molecular graphics visualization intended and used mainly to depict and explore biological macromolecule structures, such as those found in the Protein Data Bank (PDB).
The European Bioinformatics Institute (EMBL-EBI) is an intergovernmental organization (IGO) which, as part of the European Molecular Biology Laboratory (EMBL) family, focuses on research and services in bioinformatics. It is located on the Wellcome Genome Campus in Hinxton near Cambridge, and employs over 600 full-time equivalent (FTE) staff. Institute leaders such as Rolf Apweiler, Alex Bateman, Ewan Birney, and Guy Cochrane, an adviser on the National Genomics Data Center Scientific Advisory Board, serve as part of the international research network of the BIG Data Center at the Beijing Institute of Genomics.
The Protein Data Bank (PDB) file format is a textual file format describing the three-dimensional structures of molecules held in the Protein Data Bank, now succeeded by the mmCIF format. The PDB format accordingly provides for description and annotation of protein and nucleic acid structures including atomic coordinates, secondary structure assignments, as well as atomic connectivity. In addition experimental metadata are stored. The PDB format is the legacy file format for the Protein Data Bank which has kept data on biological macromolecules in the newer PDBx/mmCIF file format since 2014.
Biological data visualization is a branch of bioinformatics concerned with the application of computer graphics, scientific visualization, and information visualization to different areas of the life sciences. This includes visualization of sequences, genomes, alignments, phylogenies, macromolecular structures, systems biology, microscopy, and magnetic resonance imaging data. Software tools used for visualizing biological data range from simple, standalone programs to complex, integrated systems.
PDBsum is a database that provides an overview of the contents of each 3D macromolecular structure deposited in the Protein Data Bank (PDB).
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.

The Protein Common Interface Database (ProtCID) is a database of similar protein-protein interfaces in crystal structures of homologous proteins.
SWISS-MODEL is a structural bioinformatics web-server dedicated to homology modeling of 3D protein structures. Homology modeling is currently the most accurate method to generate reliable three-dimensional protein structure models and is routinely used in many practical applications. Homology modelling methods make use of experimental protein structures ("templates") to build models for evolutionary related proteins ("targets").
Computer Atlas of Surface Topography of Proteins (CASTp) aims to provide comprehensive and detailed quantitative characterization of topographic features of protein, is now updated to version 3.0. Since its release in 2006, the CASTp server has ≈45000 visits and fulfills ≈33000 calculation requests annually. CASTp has been proven as a confident tool for a wide range of researches, including investigations of signaling receptors, discoveries of cancer therapeutics, understanding of mechanism of drug actions, studies of immune disorder diseases, analysis of protein–nanoparticle interactions, inference of protein functions and development of high-throughput computational tools. This server is maintained by Jie Liang's lab in University of Illinois at Chicago.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.
Molecular Operating Environment (MOE) is a drug discovery software platform that integrates visualization, modeling and simulations, as well as methodology development, in one package. MOE scientific applications are used by biologists, medicinal chemists and computational chemists in pharmaceutical, biotechnology and academic research. MOE runs on Windows, Linux, Unix, and macOS. Main application areas in MOE include structure-based design, fragment-based design, ligand-based design, pharmacophore discovery, medicinal chemistry applications, biologics applications, structural biology and bioinformatics, protein and antibody modeling, molecular modeling and simulations, virtual screening, cheminformatics & QSAR. The Scientific Vector Language (SVL) is the built-in command, scripting and application development language of MOE.
References
- ↑ Laskowski, RA (2011). "Protein structure databases". Mol Biotechnol. 48 (2): 183–98. doi:10.1007/s12033-010-9372-4. PMID 21225378. S2CID 45184564.
- ↑ Berman, H. M. (January 2008). "The Protein Data Bank: a historical perspective" (PDF). Acta Crystallographica Section A. A64 (1): 88–95. doi: 10.1107/S0108767307035623 . PMID 18156675.
- ↑ Laskowski RA, Hutchinson EG, Michie AD, Wallace AC, Jones ML, Thornton JM (December 1997). "PDBsum: a Web-based database of summaries and analyses of all PDB structures". Trends Biochem. Sci. 22 (12): 488–90. doi:10.1016/S0968-0004(97)01140-7. PMID 9433130.