Nucleotides are organic molecules composed of a nitrogenous base, a pentose sugar and a phosphate. They serve as monomeric units of the nucleic acid polymers – deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), both of which are essential biomolecules within all life-forms on Earth. Nucleotides are obtained in the diet and are also synthesized from common nutrients by the liver.
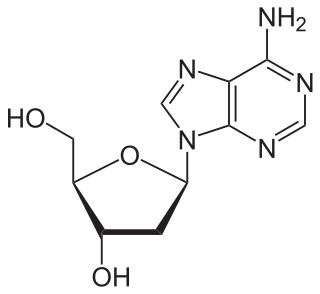
Nucleosides are glycosylamines that can be thought of as nucleotides without a phosphate group. A nucleoside consists simply of a nucleobase and a five-carbon sugar whereas a nucleotide is composed of a nucleobase, a five-carbon sugar, and one or more phosphate groups. In a nucleoside, the anomeric carbon is linked through a glycosidic bond to the N9 of a purine or the N1 of a pyrimidine. Nucleotides are the molecular building blocks of DNA and RNA.

Thymidine, also known as deoxythymidine, deoxyribosylthymine, or thymine deoxyriboside, is a pyrimidine deoxynucleoside. Deoxythymidine is the DNA nucleoside T, which pairs with deoxyadenosine (A) in double-stranded DNA. In cell biology it is used to synchronize the cells in G1/early S phase. The prefix deoxy- is often left out since there are no precursors of thymine nucleotides involved in RNA synthesis.
Zidovudine (ZDV), also known as azidothymidine (AZT), was the first antiretroviral medication used to prevent and treat HIV/AIDS. It is generally recommended for use in combination with other antiretrovirals. It may be used to prevent mother-to-child spread during birth or after a needlestick injury or other potential exposure. It is sold both by itself and together as lamivudine/zidovudine and abacavir/lamivudine/zidovudine. It can be used by mouth or by slow injection into a vein.
Reverse-transcriptase inhibitors (RTIs) are a class of antiretroviral drugs used to treat HIV infection or AIDS, and in some cases hepatitis B. RTIs inhibit activity of reverse transcriptase, a viral DNA polymerase that is required for replication of HIV and other retroviruses.

Zalcitabine, also called dideoxycytidine, is a nucleoside analog reverse-transcriptase inhibitor (NRTI) sold under the trade name Hivid. Zalcitabine was the third antiretroviral to be approved by the Food and Drug Administration (FDA) for the treatment of HIV/AIDS. It is used as part of a combination regimen.
A nucleoside triphosphate is a nucleoside containing a nitrogenous base bound to a 5-carbon sugar, with three phosphate groups bound to the sugar. They are the molecular precursors of both DNA and RNA, which are chains of nucleotides made through the processes of DNA replication and transcription. Nucleoside triphosphates also serve as a source of energy for cellular reactions and are involved in signalling pathways.

Thymidine kinase is an enzyme, a phosphotransferase : 2'-deoxythymidine kinase, ATP-thymidine 5'-phosphotransferase, EC 2.7.1.21. It can be found in most living cells. It is present in two forms in mammalian cells, TK1 and TK2. Certain viruses also have genetic information for expression of viral thymidine kinases. Thymidine kinase catalyzes the reaction:

Nucleoside analogues are structural analogues of a nucleoside, which normally contain a nucleobase and a sugar. Nucleotide analogues are analogues of a nucleotide, which normally has one to three phosphates linked to a nucleoside. Both types of compounds can deviate from what they mimick in a number of ways, as changes can be made to any of the constituent parts. They are related to nucleic acid analogues.

3′,5′-cyclic-nucleotide phosphodiesterases (EC 3.1.4.17) are a family of phosphodiesterases. Generally, these enzymes hydrolyze a nucleoside 3′,5′-cyclic phosphate to a nucleoside 5′-phosphate:

Nucleic acid metabolism is a collective term that refers to the variety of chemical reactions by which nucleic acids are either synthesized or degraded. Nucleic acids are polymers made up of a variety of monomers called nucleotides. Nucleotide synthesis is an anabolic mechanism generally involving the chemical reaction of phosphate, pentose sugar, and a nitrogenous base. Degradation of nucleic acids is a catabolic reaction and the resulting parts of the nucleotides or nucleobases can be salvaged to recreate new nucleotides. Both synthesis and degradation reactions require multiple enzymes to facilitate the event. Defects or deficiencies in these enzymes can lead to a variety of diseases.

Deoxycytidine kinase (dCK) is an enzyme which is encoded by the DCK gene in humans. dCK predominantly phosphorylates deoxycytidine (dC) and converts dC into deoxycytidine monophosphate. dCK catalyzes one of the initial steps in the nucleoside salvage pathway and has the potential to phosphorylate other preformed nucleosides, specifically deoxyadenosine (dA) and deoxyguanosine (dG), and convert them into their monophosphate forms. There has been recent biomedical research interest in investigating dCK's potential as a therapeutic target for different types of cancer.

The organic anion transporter 1 (OAT1) also known as solute carrier family 22 member 6 (SLC22A6) is a protein that in humans is encoded by the SLC22A6 gene. It is a member of the organic anion transporter (OAT) family of proteins. OAT1 is a transmembrane protein that is expressed in the brain, the placenta, the eyes, smooth muscles, and the basolateral membrane of proximal tubular cells of the kidneys. It plays a central role in renal organic anion transport. Along with OAT3, OAT1 mediates the uptake of a wide range of relatively small and hydrophilic organic anions from plasma into the cytoplasm of the proximal tubular cells of the kidneys. From there, these substrates are transported into the lumen of the nephrons of the kidneys for excretion. OAT1 homologs have been identified in rats, mice, rabbits, pigs, flounders, and nematodes.
Discovery and development of nucleoside and nucleotide reverse-transcriptase inhibitors began in the 1980s when the AIDS epidemic hit Western societies. NRTIs inhibit the reverse transcriptase (RT), an enzyme that controls the replication of the genetic material of the human immunodeficiency virus (HIV). The first NRTI was zidovudine, approved by the U.S. Food and Drug Administration (FDA) in 1987, which was the first step towards treatment of HIV. Six NRTI agents and one NtRTI have followed. The NRTIs and the NtRTI are analogues of endogenous 2´-deoxy-nucleoside and nucleotide. Drug-resistant viruses are an inevitable consequence of prolonged exposure of HIV-1 to anti-HIV drugs.
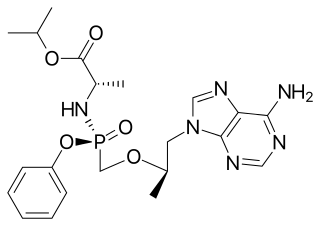
Tenofovir alafenamide, sold under the brand name Vemlidy, is an antiviral medication used against hepatitis B and HIV. It is used for the treatment of chronic hepatitis B virus (HBV) infection in adults with compensated liver disease and is given in combination with other medications for the prevention and treatment of HIV. It is taken by mouth.

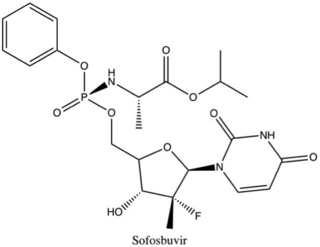
Sofosbuvir, sold under the brand name Sovaldi among others, is a medication used to treat hepatitis C. It is taken by mouth.
Janet Rideout is an organic chemist and one of the scientists who discovered that azidothymidine (AZT) could be used as an antiretroviral agent to treat Human Immunodeficiency Virus (HIV). She also played a key role in the development of acyclovir, the first effective treatment for herpes simplex virus.
Non-structural protein 5B (NS5B) inhibitors are a class of direct-acting antivirals widely used in the treatment of chronic hepatitis C. Depending on site of action and chemical composition, NS5B inhibitors may be categorized into three classes—nucleoside active site inhibitors (NIs), non-nucleoside allosteric inhibitors, and pyrophosphate analogues. Subsequently, all three classes are then subclassified. All inhibit RNA synthesis by NS5B but at different stages/sites resulting in inability of viral RNA replication. Expression of direct-acting NS5B inhibitors does not take place in cells that are not infected by hepatitis C virus, which seems to be beneficial for this class of drugs.

GS-441524 is a nucleoside analogue antiviral drug which was developed by Gilead Sciences. It is the main plasma metabolite of the antiviral prodrug remdesivir, and has a half-life of around 24 hours in human patients. Remdesivir and GS-441524 were both found to be effective in vitro against feline coronavirus strains responsible for feline infectious peritonitis (FIP), a lethal systemic disease affecting domestic cats. Remdesivir was never tested in cats, but GS-441524 has been found to be effective treatment for FIP.
Katherine Seley-Radtke is an American medicinal chemist who specializes in the discovery and design of novel nucleoside or nucleotide based enzyme inhibitors that may be used to treat infections or cancer. She has authored over 90 peer-reviewed publications, is an inventor of five issued US patents, and is a professor in the department of chemistry and biochemistry at the University of Maryland, Baltimore County. Her international impact includes scientific collaborations, policy advising and diplomatic appointments in biosecurity efforts.