
Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis.
Pyrrole is a heterocyclic, aromatic, organic compound, a five-membered ring with the formula C4H4NH. It is a colorless volatile liquid that darkens readily upon exposure to air. Substituted derivatives are also called pyrroles, e.g., N-methylpyrrole, C4H4NCH3. Porphobilinogen, a trisubstituted pyrrole, is the biosynthetic precursor to many natural products such as heme.
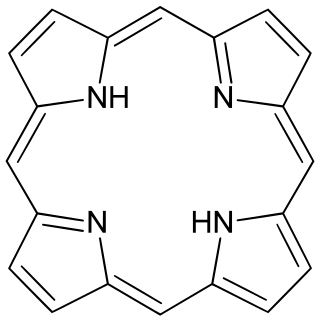
Porphyrins are a group of heterocyclic, macrocyclic, organic compounds, composed of four modified pyrrole subunits interconnected at their α carbon atoms via methine bridges. In vertebrates, an essential member of the porphyrin group is heme, which is a component of hemoproteins, whose functions include carrying oxygen in the bloodstream. In plants, an essential porphyrin derivative is chlorophyll, which is involved in light harvesting and electron transfer in photosynthesis.

In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.
The Pictet–Spengler reaction is a chemical reaction in which a β-arylethylamine undergoes condensation with an aldehyde or ketone followed by ring closure. The reaction was first discovered in 1911 by Amé Pictet and Theodor Spengler. Traditionally, an acidic catalyst in protic solvent was employed with heating; however, the reaction has been shown to work in aprotic media in superior yields and sometimes without acid catalysis. The Pictet–Spengler reaction can be considered a special case of the Mannich reaction, which follows a similar reaction pathway. The driving force for this reaction is the electrophilicity of the iminium ion generated from the condensation of the aldehyde and amine under acid conditions. This explains the need for an acid catalyst in most cases, as the imine is not electrophilic enough for ring closure but the iminium ion is capable of undergoing the reaction.
Porphine or porphin is an organic compound of empirical formula C20H14N4. It is heterocyclic and aromatic. The molecule is a flat macrocycle, consisting of four pyrrole-like rings joined by four methine bridges, which makes it the simplest of the tetrapyrroles.
The Stetter reaction is a reaction used in organic chemistry to form carbon-carbon bonds through a 1,4-addition reaction utilizing a nucleophilic catalyst. While the related 1,2-addition reaction, the benzoin condensation, was known since the 1830s, the Stetter reaction was not reported until 1973 by Dr. Hermann Stetter. The reaction provides synthetically useful 1,4-dicarbonyl compounds and related derivatives from aldehydes and Michael acceptors. Unlike 1,3-dicarbonyls, which are easily accessed through the Claisen condensation, or 1,5-dicarbonyls, which are commonly made using a Michael reaction, 1,4-dicarbonyls are challenging substrates to synthesize, yet are valuable starting materials for several organic transformations, including the Paal–Knorr synthesis of furans and pyrroles. Traditionally utilized catalysts for the Stetter reaction are thiazolium salts and cyanide anion, but more recent work toward the asymmetric Stetter reaction has found triazolium salts to be effective. The Stetter reaction is an example of umpolung chemistry, as the inherent polarity of the aldehyde is reversed by the addition of the catalyst to the aldehyde, rendering the carbon center nucleophilic rather than electrophilic.
Lanthanide triflates are triflate salts of the lanthanides. These salts have been investigated for application in organic synthesis as Lewis acid catalysts. These catalysts function similarly to aluminium chloride or ferric chloride, but they are water-tolerant (stable in water). Commonly written as Ln(OTf)3·(H2O)9 the nine waters are bound to the lanthanide, and the triflates are counteranions, so more accurately lanthanide triflate nonahydrate is written as [Ln(H2O)9](OTf)3.

Protoporphyrin IX is an organic compound, classified as a porphyrin, that plays an important role in living organisms as a precursor to other critical compounds like heme (hemoglobin) and chlorophyll. It is a deeply colored solid that is not soluble in water. The name is often abbreviated as PPIX.

The Nozaki–Hiyama–Kishi reaction is a nickel/chromium coupling reaction forming an alcohol from the reaction of an aldehyde with an allyl or vinyl halide. In their original 1977 publication, Tamejiro Hiyama and Hitoshi Nozaki reported on a chromium(II) salt solution prepared by reduction of chromic chloride by lithium aluminium hydride to which was added benzaldehyde and allyl chloride:
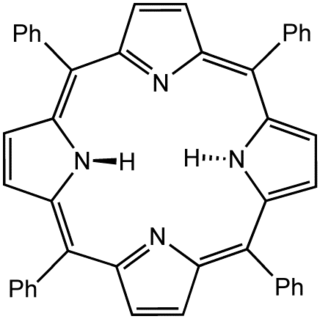
Tetraphenylporphyrin, abbreviated TPP or H2TPP, is a synthetic heterocyclic compound that resembles naturally occurring porphyrins. Porphyrins are dyes and cofactors found in hemoglobin and cytochromes and are related to chlorophyll and vitamin B12. The study of naturally occurring porphyrins is complicated by their low symmetry and the presence of polar substituents. Tetraphenylporphyrin is hydrophobic, symmetrically substituted, and easily synthesized. The compound is a dark purple solid that dissolves in nonpolar organic solvents such as chloroform and benzene.
Alcohol oxidation is a collection of oxidation reactions in organic chemistry that convert alcohols to aldehydes, ketones, carboxylic acids, and esters. The reaction mainly applies to primary and secondary alcohols. Secondary alcohols form ketones, while primary alcohols form aldehydes or carboxylic acids.
In organic chemistry, the Baylis–Hillman, Morita–Baylis–Hillman, or MBH reaction is a carbon-carbon bond-forming reaction between an activated alkene and a carbon electrophile in the presence of a nucleophilic catalyst, such as a tertiary amine or phosphine. The product is densely functionalized, joining the alkene at the α-position to a reduced form of the electrophile.
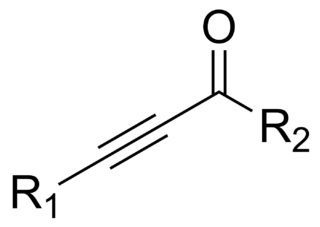
In organic chemistry, an ynone is an organic compound containing a ketone functional group and a C≡C triple bond. Many ynones are α,β-ynones, where the carbonyl and alkyne groups are conjugated. Capillin is a naturally occurring example. Some ynones are not conjugated.
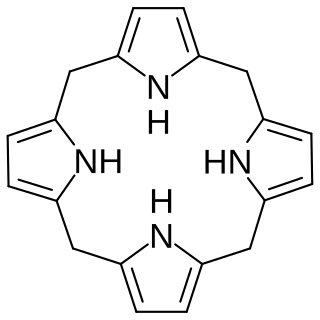
In biochemistry, a porphyrinogen is a member of a class of naturally occurring compounds with a tetrapyrrole core, a macrocycle of four pyrrole rings connected by four methylene bridges. They can be viewed as derived from the parent compound hexahydroporphine by the substitution of various functional groups for hydrogen atoms in the outermost (20-carbon) ring.
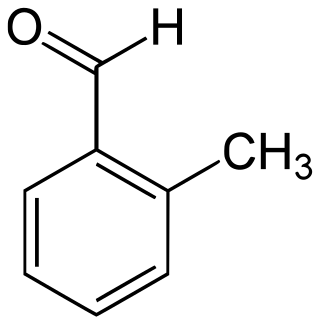
2-Methylbenzaldehyde is an organic compound with the formula CH3C6H4CHO. It is a colorless liquid.

Iron(tetraporphyrinato) chloride is the coordination complex with the formula Fe(TPP)Cl where TPP is the dianion [C44H28N4]2-. The compound forms blue microcrystals that dissolve in chlorinated solvent to give brown solutions. In terms of structure, the complex is five-coordinate with idealized C4v point group symmetry. It is one of more common transition metal porphyrin complexes.
Paul Wilhelm Karl Rothemund was a chemist who developed reactions related to porphyrins. The Rothemund reaction the still-classic process for the synthesis of these compounds is named for him. His grandson Paul W. K. Rothemund is also a chemist.

Transition metal porphyrin complexes are a family of coordination complexes of the conjugate base of porphyrins. Iron porphyrin complexes occur widely in Nature, which has stimulated extensive studies on related synthetic complexes. The metal-porphyrin interaction is a strong one such that metalloporphyrins are thermally robust. They are catalysts and exhibit rich optical properties, although these complexes remain mainly of academic interest.

Phosphorus-centered porphyrins are conjugated polycyclic ring systems consisting of either four pyrroles with inward-facing nitrogens and a phosphorus atom at their core or porphyrins with one of the four pyrroles substituted for a phosphole. Unmodified porphyrins are composed of pyrroles and linked by unsaturated hydrocarbon bridges often acting as multidentate ligands centered around a transition metal like Cu II, Zn II, Co II, Fe III. Being highly conjugated molecules with many accessible energy levels, porphyrins are used in biological systems to perform light-energy conversion and modified synthetically to perform similar functions as a photoswitch or catalytic electron carriers. Phosphorus III and V ions are much smaller than the typical metal centers and bestow distinct photochemical properties unto the porphyrin. Similar compounds with other pnictogen cores or different polycyclic rings coordinated to phosphorus result in other changes to the porphyrin’s chemistry.

