
Marfan syndrome (MFS) is a multi-systemic genetic disorder that affects the connective tissue. Those with the condition tend to be tall and thin, with long arms, legs, fingers, and toes. They also typically have exceptionally flexible joints and abnormally curved spines. The most serious complications involve the heart and aorta, with an increased risk of mitral valve prolapse and aortic aneurysm. The lungs, eyes, bones, and the covering of the spinal cord are also commonly affected. The severity of the symptoms is variable.

Ehlers–Danlos syndromes (EDS) are a group of 13 genetic connective-tissue disorders. Symptoms often include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. These may be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis. The current classification was last updated in 2017, when a number of rarer forms of EDS were added.

Noonan syndrome (NS) is a genetic disorder that may present with mildly unusual facial features, short height, congenital heart disease, bleeding problems, and skeletal malformations. Facial features include widely spaced eyes, light-colored eyes, low-set ears, a short neck, and a small lower jaw. Heart problems may include pulmonary valve stenosis. The breast bone may either protrude or be sunken, while the spine may be abnormally curved. Intelligence is often normal. Complications of NS can include leukemia.

Treacher Collins syndrome (TCS) is a genetic disorder characterized by deformities of the ears, eyes, cheekbones, and chin. The degree to which a person is affected, however, may vary from mild to severe. Complications may include breathing problems, problems seeing, cleft palate, and hearing loss. Those affected generally have normal intelligence.

Stickler syndrome is a group of rare genetic disorders affecting connective tissue, specifically collagen. Stickler syndrome is a subtype of collagenopathy, types II and XI. Stickler syndrome is characterized by distinctive facial abnormalities, ocular problems, hearing loss, and joint and skeletal problems. It was first studied and characterized by Gunnar B. Stickler in 1965.

Duane-radial ray syndrome, also known as Okihiro syndrome, is a rare autosomal dominant disorder that primarily affects the eyes and causes abnormalities of bones in the arms and hands. This disorder is considered to be a SALL4-related disorder due to the SALL4 gene mutations leading to these abnormalities. It is diagnosed by clinical findings on a physical exam as well as genetic testing and imaging. After being diagnosed, there are other evaluations that one may go through in order to determine the extent of the disease. There are various treatments for the symptoms of this disorder.
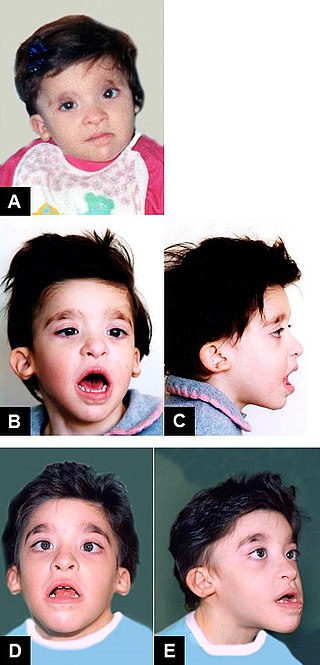
Mowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by David R. Mowat and Meredith J. Wilson in 1998. The condition affects both males and females, has been described in various countries and ethnic groups around the world, and occurs in approximately 1 in 50,000–100,000 births.

SCARF syndrome is a rare syndrome characterized by skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, psychomotor retardation, and facial abnormalities. These characteristics are what make up the acronym SCARF. It shares some features with Lenz-Majewski hyperostotic dwarfism. It is a very rare disease with an incidence rate of approximately one in a million newborns. It has been clinically described in two males who were maternal cousins, as well as a 3-month-old female. Babies affected by this syndrome tend to have very loose skin, giving them an elderly facial appearance. Possible complications include dyspnea, abdominal hernia, heart disorders, joint disorders, and dislocations of multiple joints. It is believed that this disease's inheritance is X-linked recessive.
Loeys–Dietz syndrome (LDS) is an autosomal dominant genetic connective tissue disorder. It has features similar to Marfan syndrome and Ehlers–Danlos syndrome. The disorder is marked by aneurysms in the aorta, often in children, and the aorta may also undergo sudden dissection in the weakened layers of the wall of the aorta. Aneurysms and dissections also can occur in arteries other than the aorta. Because aneurysms in children tend to rupture early, children are at greater risk for dying if the syndrome is not identified. Surgery to repair aortic aneurysms is essential for treatment.

Axenfeld–Rieger syndrome is a rare autosomal dominant disorder, which affects the development of the teeth, eyes, and abdominal region.
Aarskog–Scott syndrome (AAS) is a rare disease inherited as X-linked and characterized by short stature, facial abnormalities, skeletal and genital anomalies. This condition mainly affects males, although females may have mild features of the syndrome.

Cornelia de Lange syndrome (CdLS) is a genetic disorder. People with Cornelia de Lange syndrome experience a range of physical, cognitive, and medical challenges ranging from mild to severe. Cornelia de Lange syndrome has a widely varied phenotype, meaning people with the syndrome have varied features and challenges. The typical features of CdLS include thick or long eyebrows, a small nose, small stature, developmental delay, long or smooth philtrum, thin upper lip and downturned mouth.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.
Marshall syndrome is a genetic disorder of the connective tissue that can cause hearing loss. The three most common areas to be affected are the eyes, which are uncommonly large, joints and the mouth and facial structures. Marshall syndrome and Stickler syndrome closely resemble each other; in fact they are so similar, some say they are the same. The condition is named for D. Weber.
Congenital generalized lipodystrophy is an extremely rare autosomal recessive condition, characterized by an extreme scarcity of fat in the subcutaneous tissues. It is a type of lipodystrophy disorder where the magnitude of fat loss determines the severity of metabolic complications. Only 250 cases of the condition have been reported, and it is estimated that it occurs in 1 in 10 million people worldwide.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, moderate to severe intellectual disability, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Epilepsy often occurs in Pitt-Hopkins. It is part of the clinical spectrum of Rett-like syndromes. Pitt-Hopkins syndrome is clinically similar to Angelman syndrome, Rett-syndrome, Mowat Wilson syndrome, and ATR-X syndrome.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.
Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are. The term progeroid syndrome does not necessarily imply progeria, which is a specific type of progeroid syndrome.
Lateral meningocele syndrome, also known as Lehman syndrome, is a very rare skeletal disorder with facial anomalies, hypotonia, and meningocele-related neurologic dysfunction. These protrusions form from membranes surrounding the spinal cord in gaps in the spine (vertebrae). They most often occur in the lower spine and damage the surrounding nerves that spread throughout the rest of the body. Examples of resulting damages are bladder function, prickling or tingling sensations, stiffness and weakness in the legs, and back pain. People affected with lateral meningocele typically have high arched eyebrows, widely spaced eyes, droopy eyes, and other facial features. There have been only 14 reported individuals with lateral meningocele syndrome with 7 of those who have a molecularly confirmed diagnosis. There is no specific treatment for this syndrome, but only supportive management including lateral spinal meningoceles, psychomotor development, musculoskeletal, and routine management.
Curry–Jones syndrome is a rare genetic disorder characterized by congenital brain, osseous, cutaneous, ocular, and intestinal anomalies.
