Related Research Articles

Fragile X syndrome (FXS) is a genetic disorder characterized by mild-to-moderate intellectual disability. The average IQ in males with FXS is under 55, while about two thirds of affected females are intellectually disabled. Physical features may include a long and narrow face, large ears, flexible fingers, and large testicles. About a third of those affected have features of autism such as problems with social interactions and delayed speech. Hyperactivity is common, and seizures occur in about 10%. Males are usually more affected than females.
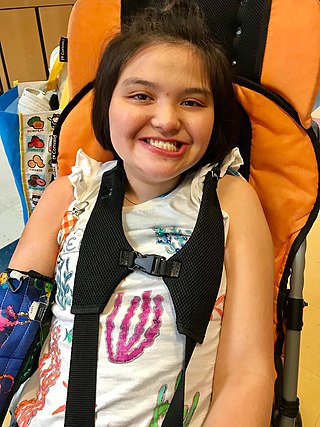
Rett syndrome (RTT) is a genetic disorder that typically becomes apparent after 6–18 months of age and almost exclusively in females. Symptoms include impairments in language and coordination, and repetitive movements. Those affected often have slower growth, difficulty walking, and a smaller head size. Complications of Rett syndrome can include seizures, scoliosis, and sleeping problems. The severity of the condition is variable.

Megalencephaly is a growth development disorder in which the brain is abnormally large. It is characterized by a brain with an average weight that is 2.5 standard deviations above the mean of the general population. Approximately 1 out of 50 children (2%) are said to have the characteristics of megalencephaly in the general population.

Polymicrogyria (PMG) is a condition that affects the development of the human brain by multiple small gyri (microgyri) creating excessive folding of the brain leading to an abnormally thick cortex. This abnormality can affect either one region of the brain or multiple regions.

Coffin–Lowry syndrome is a genetic disorder that is X-linked dominant and which causes severe mental problems sometimes associated with abnormalities of growth, cardiac abnormalities, kyphoscoliosis, as well as auditory and visual abnormalities.

Gerstmann–Sträussler–Scheinker syndrome (GSS) is an extremely rare, always fatal neurodegenerative disease that affects patients from 20 to 60 years in age. It is exclusively heritable, and is found in only a few families all over the world. It is, however, classified with the transmissible spongiform encephalopathies (TSE) due to the causative role played by PRNP, the human prion protein. GSS was first reported by the Austrian physicians Josef Gerstmann, Ernst Sträussler and Ilya Scheinker in 1936.
Neurodevelopmental disorders are a group of conditions that begin to emerge during childhood. According to the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, (DSM-5) published in 2013, these conditions generally appear in early childhood, usually before children start school, and can persist into adulthood. The key characteristic of all these disorders is that they negatively impact a person's functioning in one or more domains of life depending on the disorder and deficits it has caused. All of these disorders and their levels of impairment exist on a spectrum, and affected individuals can experience varying degrees of symptoms and deficits, despite having the same diagnosis.

Duane-radial ray syndrome, also known as Okihiro syndrome, is a rare autosomal dominant disorder that primarily affects the eyes and causes abnormalities of bones in the arms and hands. This disorder is considered to be a SALL4-related disorder due to the SALL4 gene mutations leading to these abnormalities. It is diagnosed by clinical findings on a physical exam as well as genetic testing and imaging. After being diagnosed, there are other evaluations that one may go through in order to determine the extent of the disease. There are various treatments for the symptoms of this disorder.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.
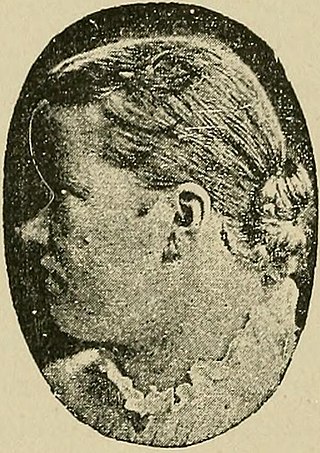
Muenke syndrome, also known as FGFR3-related craniosynostosis, is a human specific condition characterized by the premature closure of certain bones of the skull during development, which affects the shape of the head and face. First described by Maximilian Muenke, the syndrome occurs in about 1 in 30,000 newborns. This condition accounts for an estimated 8 percent of all cases of craniosynostosis.

Dubowitz syndrome is a rare genetic disorder characterized by microcephaly, stunted growth, and a receding chin. Symptoms vary among patients, but other characteristics include a soft, high-pitched voice, partial webbing of the fingers and toes, palate deformations, genital abnormalities, language difficulties, and an aversion to crowds. The pathogenesis of the disease is yet to be identified, and no medical tests can definitively diagnose the disease. The primary method of diagnosis is to identify facial phenotypes. Since it was first described in 1965 by English physician Victor Dubowitz, over 140 cases have been reported worldwide. Although the majority of cases have been reported from the United States, Germany, and Russia, the disorder appears to affect both genders and all ethnicities equally.

Allan–Herndon–Dudley syndrome is a rare X-linked inherited disorder of brain development that causes both moderate to severe intellectual disability and problems with speech and movement.

Synaptotagmin-1 is a protein that in humans is encoded by the SYT1 gene.
Ohtahara syndrome (OS), also known as early infantile epileptic encephalopathy (EIEE) is a progressive epileptic encephalopathy. The syndrome is outwardly characterized by tonic spasms and partial seizures within the first few months of life, and receives its more elaborate name from the pattern of burst activity on an electroencephalogram (EEG). It is an extremely debilitating progressive neurological disorder, involving intractable seizures and severe intellectual disabilities. No single cause has been identified, although in many cases structural brain damage is present.

Angelman syndrome or Angelman's syndrome (AS) is a genetic disorder that mainly affects the nervous system. Symptoms include a small head and a specific facial appearance, severe intellectual disability, developmental disability, limited to no functional speech, balance and movement problems, seizures, and sleep problems. Children usually have a happy personality and have a particular interest in water. The symptoms generally become noticeable by one year of age.
EAST syndrome is a syndrome consisting of epilepsy, ataxia, sensorineural deafness and salt-wasting renal tubulopathy. The tubulopathy in this condition predispose to hypokalemic metabolic alkalosis with normal blood pressure. Hypomagnesemia may also be present.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, epilepsy, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Pitt–Hopkins syndrome can be marked by intellectual disabilities as well as problems with socializing. It is part of the clinical spectrum of Rett-like syndromes.
Kohlschütter-Tönz syndrome (KTS), also called amelo-cerebro-hypohidrotic syndrome, is a rare inherited syndrome characterized by epilepsy, psychomotor delay or regression, intellectual disability, and yellow teeth caused by amelogenesis imperfecta. It is a type A ectodermal dysplasia.
Burnside–Butler syndrome is a name that has been applied to the effects of microdeletion of DNA sequences involving four neurodevelopmental genes. Varying developmental and psychiatric disorders have been attributed to the microdeletion; however, the great majority of people with the deletion do not have any clinical features associated with it. More studies are needed to delineate the range of clinical presentation.

Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
References
- 1 2 3 4 5 Baker K, Gordon SL, Melland H, Bumbak F, Scott DJ, Jiang TJ, et al. (September 2018). "SYT1-associated neurodevelopmental disorder: a case series". Brain. 141 (9): 2576–2591. doi: 10.1093/brain/awy209 . PMC 6113648 . PMID 30107533.
- ↑ Baker K, Gordon SL, Grozeva D, van Kogelenberg M, Roberts NY, Pike M, et al. (April 2015). "Identification of a human synaptotagmin-1 mutation that perturbs synaptic vesicle cycling". The Journal of Clinical Investigation. 125 (4): 1670–8. doi: 10.1172/JCI79765 . PMC 4396464 . PMID 25705886.
- ↑ "OMIM entry: Baker-Gordon Syndrome".