Diagnosis
| | This section is empty. You can help by adding to it. (February 2018) |
| Saal Bulas syndrome | |
|---|---|
| Named after |
|
Saal Bulas syndrome is listed as a "rare disease" by the Office of Rare Diseases (ORD) of the National Institutes of Health (NIH). This means that Saal Bulas syndrome, or a subtype of Saal Bulas syndrome, affects fewer than 200,000 people in the US population.
This syndrome consists of ectrodactyly or lobster-like hands, diaphragmatic hernia and absence of the corpus callosum. [1]
In addition to these the following problems may also be present.
| | This section is empty. You can help by adding to it. (February 2018) |
| | This section is empty. You can help by adding to it. (February 2018) |
The syndrome was first described by American paediatricians Howard M. Saal and Dorothy I. Bulas in 1995. [2]

Congenital diaphragmatic hernia (CDH) is a birth defect of the diaphragm. The most common type of CDH is a Bochdalek hernia; other types include Morgagni hernia, diaphragm eventration and central tendon defects of the diaphragm. Malformation of the diaphragm allows the abdominal organs to push into the chest cavity, hindering proper lung formation.
Agenesis of the corpus callosum (ACC) is a rare birth defect in which there is a complete or partial absence of the corpus callosum. It occurs when the development of the corpus callosum, the band of white matter connecting the two hemispheres in the brain, in the embryo is disrupted. The result of this is that the fibers that would otherwise form the corpus callosum are instead longitudinally oriented along the ipsilateral ventricular wall and form structures called Probst bundles.

Hypoplasia is underdevelopment or incomplete development of a tissue or organ. Although the term is not always used precisely, it properly refers to an inadequate or below-normal number of cells. Hypoplasia is similar to aplasia, but less severe. It is technically not the opposite of hyperplasia. Hypoplasia is a congenital condition, while hyperplasia generally refers to excessive cell growth later in life.
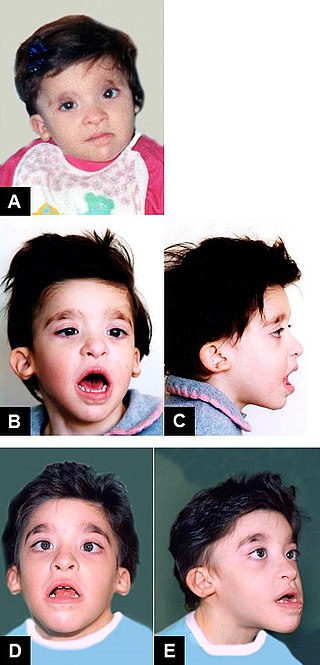
Mowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by David R. Mowat and Meredith J. Wilson in 1998. The condition affects both males and females, has been described in various countries and ethnic groups around the world, and occurs in approximately 1 in 50,000–100,000 births.
Saal may refer to:
The Fetal Treatment Center at the University of California, San Francisco is a multidisciplinary care center dedicated to the diagnosis, treatment, and long-term follow-up of fetal birth defects. It combines the talents of specialists in pediatric surgery, genetics, obstetrics/perinatology, radiology, nursing, and neonatal medicine.
Shapiro syndrome is an extremely rare disorder consisting of paroxysmal hypothermia, hyperhydrosis (sweating), and agenesis of the corpus callosum with onset typically on adulthood. The disease affects about 50 people worldwide. The duration and frequency of the episodes vary from person to person, with some episodes lasting hours to weeks and occurring from hours to years. Very little is known about the disease due to the small number of people affected.

Acrocallosal syndrome is an extremely rare autosomal recessive syndrome characterized by corpus callosum agenesis, polydactyly, multiple dysmorphic features, motor and intellectual disabilities, and other symptoms. The syndrome was first described by Albert Schinzel in 1979. Mutations in KIF7 are causative for ACLS, and mutations in GLI3 are associated with a similar syndrome.

Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome that is usually lethal in the neonatal period. Fryns (1987) reviewed the syndrome.
Donnai–Barrow syndrome is a genetic disorder first described by Dian Donnai and Margaret Barrow in 1993. It is associated with LRP2. It is an inherited (genetic) disorder that affects many parts of the body.
Toriello–Carey syndrome is a genetic disorder that is characterized by Pierre Robin sequence and agenesis of the corpus callosum. Children with the disorder also possess a characteristic facial phenotype.

Pai syndrome, also known as Median cleft of the upper lip-corpus callosum lipoma-midline facial cutaneous polyps syndrome, is a very rare genetic disorder which is characterized by nervous system, cutaneous, ocular, nasal and bucal anomalies with facial dysmorphisms.
Proud syndrome is a very rare genetic disorder which is characterized by severe intellectual disabilities, corpus callosum agenesis, epilepsy, and spasticity. It is a type of syndromic X-linked intellectual disability.
Meacham syndrome is a rare genetic disorder which is characterized by lung, diaphragmatic and genitourinary anomalies.
Holoprosencephaly-ectrodactyly-cleft lip/palate syndrome, also simply known as Hartsfield syndrome, is a rare genetic disorder characterized by the presence of variable holoprosencephaly, ectrodactyly, cleft lip and palate, alongside generalized ectodermal abnormalities. Additional findings include endocrine anomalies and developmental delays.
| | This genetic disorder article is a stub. You can help Wikipedia by expanding it. |