Related Research Articles

A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Ehlers–Danlos syndromes are a group of rare genetic connective tissue disorders. Symptoms may include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. These can be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis.

Weissenbacher–Zweymuller syndrome (WZS), also called Pierre-Robin syndrome with fetal chondrodysplasia, is an autosomal recessive congenital disorder, linked to mutations in the COL11A2 gene, which codes for the α2 strand of collagen type XI. It is a collagenopathy, types II and XI disorder.

Autoimmune polyendocrine syndromes (APSs), also called polyglandular autoimmune syndromes (PGASs) or polyendocrine autoimmune syndromes (PASs), are a heterogeneous group of rare diseases characterized by autoimmune activity against more than one endocrine organ, although non-endocrine organs can be affected. There are three types of APS, and there are a number of other diseases which involve endocrine autoimmunity.
Niemann–Pick disease is a group of severe inherited metabolic disorders, in which sphingomyelin accumulates in lysosomes in cells.

Oculopharyngeal muscular dystrophy (OPMD) is a rare form of muscular dystrophy with symptoms generally starting when an individual is 40 to 50 years old. It can be autosomal dominant neuromuscular disease or autosomal recessive. The most common inheritance of OPMD is autosomal dominant, which means only one copy of the mutated gene needs to be present in each cell. Children of an affected parent have a 50% chance of inheriting the mutant gene.

Horner's syndrome, also known as oculosympathetic paresis, is a combination of symptoms that arises when a group of nerves known as the sympathetic trunk is damaged. The signs and symptoms occur on the same side (ipsilateral) as it is a lesion of the sympathetic trunk. It is characterized by miosis, partial ptosis, apparent anhydrosis, with apparent enophthalmos.

Mulibrey nanism, is a rare autosomal recessive congenital disorder. It causes severe growth failure along with abnormalities of the heart, muscle, liver, brain and eye. TRIM37 is responsible for various cellular functions including developmental patterning.
Infantile neuronal ceroid lipofuscinoses (INCL) or Santavuori disease or Hagberg-Santavuori disease or Santavuori-Haltia disease or Infantile Finnish type neuronal ceroid lipofuscinosis or Balkan disease is a form of NCL and inherited as a recessive autosomal genetic trait. The disorder is progressive, degenerative and fatal, extremely rare worldwide – with approximately 60 official cases reported by 1982, perhaps 100 sufferers in total today – but relatively common in Finland due to the local founder effect.
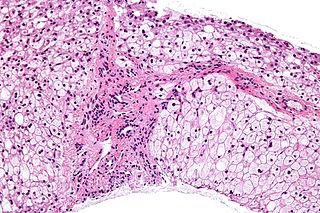
Glycogen storage disease type III is an autosomal recessive metabolic disorder and inborn error of metabolism characterized by a deficiency in glycogen debranching enzymes.
Jackson–Weiss syndrome (JWS) is a genetic disorder characterized by foot abnormalities and the premature fusion of certain bones of the skull (craniosynostosis), which prevents further growth of the skull and affects the shape of the head and face. This genetic disorder can also sometimes cause intellectual disability and crossed eyes. It was characterized in 1976.
Bannayan–Riley–Ruvalcaba syndrome (BRRS) is a rare overgrowth syndrome and hamartomatous disorder with occurrence of multiple subcutaneous lipomas, macrocephaly and hemangiomas. The disease is inherited in an autosomal dominant manner. The disease belongs to a family of hamartomatous polyposis syndromes, which also includes Peutz–Jeghers syndrome, juvenile polyposis and Cowden syndrome. Mutation of the PTEN gene underlies this syndrome, as well as Cowden syndrome, Proteus syndrome, and Proteus-like syndrome, these four syndromes are referred to as PTEN Hamartoma-Tumor Syndromes.

Oculocerebrorenal syndrome is a rare X-linked recessive disorder characterized by congenital cataracts, hypotonia, intellectual disability, proximal tubular acidosis, aminoaciduria and low-molecular-weight proteinuria. Lowe syndrome can be considered a cause of Fanconi syndrome.
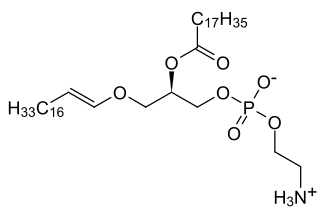
Rhizomelic chondrodysplasia punctata is a rare developmental brain disorder characterized by systemic shortening of the proximal bones, seizures, recurrent respiratory tract infections and congenital cataracts. The affected individuals have low levels of plasmalogens.

Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The FAH gene is thought to be involved in the catabolism of the amino acid phenylalanine in humans.

Acrocallosal syndrome is a rare autosomal recessive syndrome characterized by corpus callosum agenesis, polydactyly, multiple dysmorphic features, motor and intellectual disabilities, and other symptoms. The syndrome was first described by Albert Schinzel in 1979.
Autoimmune polyendocrine syndrome type 1 (APS-1), is a subtype of autoimmune polyendocrine syndrome in which multiple endocrine glands dysfunction as a result of autoimmunity. It is a genetic disorder inherited in autosomal recessive fashion due to a defect in the AIRE gene , which is located on chromosome 21 and normally confers immune tolerance.
A Finnish heritage disease is a genetic disease or disorder that is significantly more common in people whose ancestors were ethnic Finns, natives of Finland and Sweden (Meänmaa) and Russia. There are 36 rare diseases regarded as Finnish heritage diseases. The diseases are not restricted to Finns; they are genetic diseases with far wider distribution in the world, but due to founder effects and genetic isolation they are more common in Finns.
Albert Fredrik de la Chapelle, MD, Ph.D was a Finnish human geneticist, long-time head of Finland's first Department of Medical Genetics at the University of Helsinki, and subsequently professor of Human Cancer Genetics at Ohio State University. He was best known for his role in the elucidation of the genetics of hereditary colorectal cancer and Lynch syndrome.
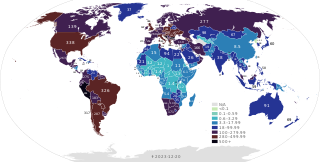
This article provides a general overview and documents the status of locations affected by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus which causes coronavirus disease 2019 (COVID-19) and is responsible for the COVID-19 pandemic. The first human cases of COVID-19 were identified in Wuhan, the capital of the province of Hubei in China in December 2019. The most recent country or territory to report its first confirmed case was the Federated States of Micronesia on 8 January 2021.
References
- 1 2 3 Hersh, Craig P.; De Meo, Dawn L.; Silverman, Edwin K. (2005). "10. Chronic obstructive pulmonary disease". In Lomas, David; Silverman, Edwin; Weiss, Scott; Shapiro, Steven (eds.). Respiratory Genetics. Hodder Arnold. p. 265. ISBN 0-340-814322.
- ↑ "Sialuria: MedlinePlus Genetics". medlineplus.gov. Retrieved 10 January 2021.
- ↑ "Sialuria, French type | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 10 January 2021.
- ↑ "Orphanet: Sialuria". www.orpha.net. Retrieved 10 January 2021.