
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.
Protein engineering is the process of developing useful or valuable proteins through the design and production of unnatural polypeptides, often by altering amino acid sequences found in nature. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; it is important in medicine and biotechnology.
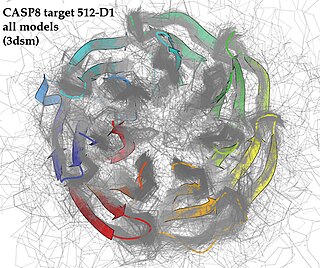
Critical Assessment of Structure Prediction (CASP), sometimes called Critical Assessment of Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and software users. Even though the primary goal of CASP is to help advance the methods of identifying protein three-dimensional structure from its amino acid sequence many view the experiment more as a “world championship” in this field of science. More than 100 research groups from all over the world participate in CASP on a regular basis and it is not uncommon for entire groups to suspend their other research for months while they focus on getting their servers ready for the experiment and on performing the detailed predictions.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
This list of structural comparison and alignment software is a compilation of software tools and web portals used in pairwise or multiple structural comparison and structural alignment.

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein. Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of a sequence alignment that maps residues in the query sequence to residues in the template sequence. It has been seen that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.
Loop modeling is a problem in protein structure prediction requiring the prediction of the conformations of loop regions in proteins with or without the use of a structural template. Computer programs that solve these problems have been used to research a broad range of scientific topics from ADP to breast cancer. Because protein function is determined by its shape and the physiochemical properties of its exposed surface, it is important to create an accurate model for protein/ligand interaction studies. The problem arises often in homology modeling, where the tertiary structure of an amino acid sequence is predicted based on a sequence alignment to a template, or a second sequence whose structure is known. Because loops have highly variable sequences even within a given structural motif or protein fold, they often correspond to unaligned regions in sequence alignments; they also tend to be located at the solvent-exposed surface of globular proteins and thus are more conformationally flexible. Consequently, they often cannot be modeled using standard homology modeling techniques. More constrained versions of loop modeling are also used in the data fitting stages of solving a protein structure by X-ray crystallography, because loops can correspond to regions of low electron density and are therefore difficult to resolve.
In computational biology, de novo protein structure prediction refers to an algorithmic process by which protein tertiary structure is predicted from its amino acid primary sequence. The problem itself has occupied leading scientists for decades while still remaining unsolved. According to Science, the problem remains one of the top 125 outstanding issues in modern science. At present, some of the most successful methods have a reasonable probability of predicting the folds of small, single-domain proteins within 1.5 angstroms over the entire structure.
RAPTOR is protein threading software used for protein structure prediction. It has been replaced by RaptorX, which is much more accurate than RAPTOR.
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.
RaptorX is a software and web server for protein structure and function prediction that is free for non-commercial use. RaptorX is among the most popular methods for protein structure prediction. Like other remote homology recognition/protein threading techniques, RaptorX is able to regularly generate reliable protein models when the widely used PSI-BLAST cannot. However, RaptorX is also significantly different from those profile-based methods in that RaptorX excels at modeling of protein sequences without a large number of sequence homologs by exploiting structure information. RaptorX Server has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods.

David Tudor Jones is a Professor of Bioinformatics, and Head of Bioinformatics Group in the University College London. He is also the director in Bloomsbury Center for Bioinformatics, which is a joint Research Centre between UCL and Birkbeck, University of London and which also provides bioinformatics training and support services to biomedical researchers. In 2013, he is a member of editorial boards for PLoS ONE, BioData Mining, Advanced Bioinformatics, Chemical Biology & Drug Design, and Protein: Structure, Function and Bioinformatics.
Swiss-model is a structural bioinformatics web-server dedicated to homology modeling of 3D protein structures. As of 2024, homology modeling is the most accurate method to generate reliable three-dimensional protein structure models and is routinely used in many practical applications. Homology modelling methods make use of experimental protein structures (templates) to build models for evolutionary related proteins (targets).
The HH-suite is an open-source software package for sensitive protein sequence searching. It contains programs that can search for similar protein sequences in protein sequence databases. Sequence searches are a standard tool in modern biology with which the function of unknown proteins can be inferred from the functions of proteins with similar sequences. HHsearch and HHblits are two main programs in the package and the entry point to its search function, the latter being a faster iteration. HHpred is an online server for protein structure prediction that uses homology information from HH-suite.
A protein superfamily is the largest grouping (clade) of proteins for which common ancestry can be inferred. Usually this common ancestry is inferred from structural alignment and mechanistic similarity, even if no sequence similarity is evident. Sequence homology can then be deduced even if not apparent. Superfamilies typically contain several protein families which show sequence similarity within each family. The term protein clan is commonly used for protease and glycosyl hydrolases superfamilies based on the MEROPS and CAZy classification systems.

CS23D is a web server to generate 3D structural models from NMR chemical shifts. CS23D combines maximal fragment assembly with chemical shift threading, de novo structure generation, chemical shift-based torsion angle prediction, and chemical shift refinement. CS23D makes use of RefDB and ShiftX.

I-TASSER is a bioinformatics method for predicting three-dimensional structure model of protein molecules from amino acid sequences. It detects structure templates from the Protein Data Bank by a technique called fold recognition. The full-length structure models are constructed by reassembling structural fragments from threading templates using replica exchange Monte Carlo simulations. I-TASSER is one of the most successful protein structure prediction methods in the community-wide CASP experiments.
Non-coding RNAs have been discovered using both experimental and bioinformatic approaches. Bioinformatic approaches can be divided into three main categories. The first involves homology search, although these techniques are by definition unable to find new classes of ncRNAs. The second category includes algorithms designed to discover specific types of ncRNAs that have similar properties. Finally, some discovery methods are based on very general properties of RNA, and are thus able to discover entirely new kinds of ncRNAs.