In chemistry, dehydrogenation is a chemical reaction that involves the removal of hydrogen, usually from an organic molecule. It is the reverse of hydrogenation. Dehydrogenation is important, both as a useful reaction and a serious problem. At its simplest, it's a useful way of converting alkanes, which are relatively inert and thus low-valued, to olefins, which are reactive and thus more valuable. Alkenes are precursors to aldehydes, alcohols, polymers, and aromatics. As a problematic reaction, the fouling and inactivation of many catalysts arises via coking, which is the dehydrogenative polymerization of organic substrates.
The Heck reaction is the chemical reaction of an unsaturated halide with an alkene in the presence of a base and a palladium catalyst to form a substituted alkene. It is named after Tsutomu Mizoroki and Richard F. Heck. Heck was awarded the 2010 Nobel Prize in Chemistry, which he shared with Ei-ichi Negishi and Akira Suzuki, for the discovery and development of this reaction. This reaction was the first example of a carbon-carbon bond-forming reaction that followed a Pd(0)/Pd(II) catalytic cycle, the same catalytic cycle that is seen in other Pd(0)-catalyzed cross-coupling reactions. The Heck reaction is a way to substitute alkenes.
The Stille reaction is a chemical reaction widely used in organic synthesis. The reaction involves the coupling of two organic groups, one of which is carried as an organotin compound (also known as organostannanes). A variety of organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.
The Suzuki reaction is an organic reaction, classified as a cross-coupling reaction, where the coupling partners are a boronic acid and an organohalide and the catalyst is a palladium(0) complex. It was first published in 1979 by Akira Suzuki, and he shared the 2010 Nobel Prize in Chemistry with Richard F. Heck and Ei-ichi Negishi for their contribution to the discovery and development of palladium-catalyzed cross-couplings in organic synthesis. This reaction is also known as the Suzuki–Miyaura reaction or simply as the Suzuki coupling. It is widely used to synthesize polyolefins, styrenes, and substituted biphenyls. Several reviews have been published describing advancements and the development of the Suzuki reaction. The general scheme for the Suzuki reaction is shown below, where a carbon-carbon single bond is formed by coupling a halide (R1-X) with an organoboron species (R2-BY2) using a palladium catalyst and a base. The organoboron species is usually synthesized by hydroboration or carboboration, allowing for rapid generation of molecular complexity.
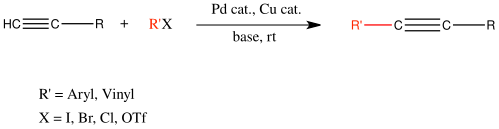
The Sonogashira reaction is a cross-coupling reaction used in organic synthesis to form carbon–carbon bonds. It employs a palladium catalyst as well as copper co-catalyst to form a carbon–carbon bond between a terminal alkyne and an aryl or vinyl halide.
The Ullmann reaction or Ullmann coupling, named after Fritz Ullmann, couples two aryl or alkyl groups with the help of copper. The reaction was first reported by Ullmann and his student Bielecki in 1901. It has been later shown that palladium and nickel can also be effectively used.
The Negishi coupling is a widely employed transition metal catalyzed cross-coupling reaction. The reaction couples organic halides or triflates with organozinc compounds, forming carbon-carbon bonds (C-C) in the process. A palladium (0) species is generally utilized as the metal catalyst, though nickel is sometimes used. A variety of nickel catalysts in either Ni0 or NiII oxidation state can be employed in Negishi cross couplings such as Ni(PPh3)4, Ni(acac)2, Ni(COD)2 etc.
In organic chemistry, carbon–hydrogen bond functionalization is a type of organic reaction in which a carbon–hydrogen bond is cleaved and replaced with a C−X bond. The term usually implies that a transition metal is involved in the C−H cleavage process. Reactions classified by the term typically involve the hydrocarbon first to react with a metal catalyst to create an organometallic complex in which the hydrocarbon is coordinated to the inner-sphere of a metal, either via an intermediate "alkane or arene complex" or as a transition state leading to a "M−C" intermediate. The intermediate of this first step can then undergo subsequent reactions to produce the functionalized product. Important to this definition is the requirement that during the C−H cleavage event, the hydrocarbyl species remains associated in the inner-sphere and under the influence of "M".
In organic chemistry, the Buchwald–Hartwig amination is a chemical reaction for the synthesis of carbon–nitrogen bonds via the palladium-catalyzed coupling reactions of amines with aryl halides. Although Pd-catalyzed C–N couplings were reported as early as 1983, Stephen L. Buchwald and John F. Hartwig have been credited, whose publications between 1994 and the late 2000s established the scope of the transformation. The reaction's synthetic utility stems primarily from the shortcomings of typical methods for the synthesis of aromatic C−N bonds, with most methods suffering from limited substrate scope and functional group tolerance. The development of the Buchwald–Hartwig reaction allowed for the facile synthesis of aryl amines, replacing to an extent harsher methods while significantly expanding the repertoire of possible C−N bond formations.
In organic chemistry, the Kumada coupling is a type of cross coupling reaction, useful for generating carbon–carbon bonds by the reaction of a Grignard reagent and an organic halide. The procedure uses transition metal catalysts, typically nickel or palladium, to couple a combination of two alkyl, aryl or vinyl groups. The groups of Robert Corriu and Makoto Kumada reported the reaction independently in 1972.
Metal carbon dioxide complexes are coordination complexes that contain carbon dioxide ligands. Aside from the fundamental interest in the coordination chemistry of simple molecules, studies in this field are motivated by the possibility that transition metals might catalyze useful transformations of CO2. This research is relevant both to organic synthesis and to the production of "solar fuels" that would avoid the use of petroleum-based fuels.

PEPPSI is an abbreviation for pyridine-enhanced precatalyst preparation stabilization and initiation. It refers to a family of commercially available palladium catalysts developed around 2005 by Prof. Michael G. Organ and co-workers at York University, which can accelerate various carbon-carbon and carbon-heteroatom bond forming cross-coupling reactions. In comparison to many alternative palladium catalysts, Pd-PEPPSI-type complexes are stable to air and moisture and are relatively easy to synthesize and handle.

A metal-phosphine complex is a coordination complex containing one or more phosphine ligands. Almost always, the phosphine is an organophosphine of the type R3P (R = alkyl, aryl). Metal phosphine complexes are useful in homogeneous catalysis. Prominent examples of metal phosphine complexes include Wilkinson's catalyst (Rh(PPh3)3Cl), Grubbs' catalyst, and tetrakis(triphenylphosphine)palladium(0).
Decarboxylative cross coupling reactions are chemical reactions in which a carboxylic acid is reacted with an organic halide to form a new carbon-carbon bond, concomitant with loss of CO2. Aryl and alkyl halides participate. Metal catalyst, base, and oxidant are required.

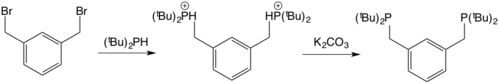
Phosphinooxazolines are a class of chiral ligands used in asymmetric catalysis. Their complexes are particularly effective at generating single enatiomers in reactions involving highly symmetric transition states, such as allylic substitutions, which are typically difficult to perform stereoselectively. The ligands are bidentate and have been shown to be hemilabile with the softer P‑donor being more firmly bound than the harder N‑donor.

In organometallic chemistry, palladium-NHC complexes are a family of organopalladium compounds in which palladium forms a coordination complex with N-heterocyclic carbenes (NHCs). They have been investigated for applications in homogeneous catalysis, particularly cross-coupling reactions.
Karen Ila Goldberg is an American chemist, currently the Vagelos Professor of Energy Research at University of Pennsylvania. Goldberg is most known for her work in inorganic and organometallic chemistry. Her most recent research focuses on catalysis, particularly on developing catalysts for oxidation, as well as the synthesis and activation of molecular oxygen. In 2018, Goldberg was elected to the National Academy of Sciences.
Dialkylbiaryl phosphine ligands are phosphine ligands that are used in homogeneous catalysis. They have proved useful in Buchwald-Hartwig amination and etherification reactions as well as Negishi cross-coupling, Suzuki-Miyaura cross-coupling, and related reactions. In addition to these Pd-based processes, their use has also been extended to transformations catalyzed by nickel, gold, silver, copper, rhodium, and ruthenium, among other transition metals.
Palladacycle, as a class of metallacycles, refers to complexes containing at least one carbon-palladium bond. Palladacycles are invoked as intermediates in catalytic or palladium mediated reactions. They have been investigated as pre-catalysts for homogeneous catalysis and synthesis.
Heterobimetallic catalysis is an approach to catalysis that employs two different metals to promote a chemical reaction. Included in this definition are cases where: 1) each metal activates a different substrate, 2) both metals interact with the same substrate, and 3) only one metal directly interacts with the substrate(s), while the second metal interacts with the first.