
Legg–Calvé–Perthes disease (LCPD) is a childhood hip disorder initiated by a disruption of blood flow to the head of the femur. Due to the lack of blood flow, the bone dies and stops growing. Over time, healing occurs by new blood vessels infiltrating the dead bone and removing the necrotic bone which leads to a loss of bone mass and a weakening of the femoral head.

A bone tumor is an abnormal growth of tissue in bone, traditionally classified as noncancerous (benign) or cancerous (malignant). Cancerous bone tumors usually originate from a cancer in another part of the body such as from lung, breast, thyroid, kidney and prostate. There may be a lump, pain, or neurological signs from pressure. A bone tumor might present with a pathologic fracture. Other symptoms may include fatigue, fever, weight loss, anemia and nausea. Sometimes there are no symptoms and the tumour is found when investigating another problem.
The ankle, or the talocrural region, or the jumping bone (informal) is the area where the foot and the leg meet. The ankle includes three joints: the ankle joint proper or talocrural joint, the subtalar joint, and the inferior tibiofibular joint. The movements produced at this joint are dorsiflexion and plantarflexion of the foot. In common usage, the term ankle refers exclusively to the ankle region. In medical terminology, "ankle" can refer broadly to the region or specifically to the talocrural joint.
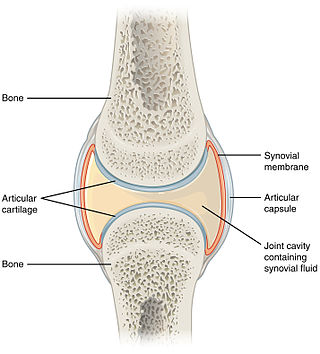
A synovial joint, also known as diarthrosis, joins bones or cartilage with a fibrous joint capsule that is continuous with the periosteum of the joined bones, constitutes the outer boundary of a synovial cavity, and surrounds the bones' articulating surfaces. This joint unites long bones and permits free bone movement and greater mobility. The synovial cavity/joint is filled with synovial fluid. The joint capsule is made up of an outer layer of fibrous membrane, which keeps the bones together structurally, and an inner layer, the synovial membrane, which seals in the synovial fluid.

Hereditary multiple osteochondromas (HMO), also known as hereditary multiple exostoses, is a disorder characterized by the development of multiple benign osteocartilaginous masses (exostoses) in relation to the ends of long bones of the lower limbs such as the femurs and tibias and of the upper limbs such as the humeri and forearm bones. They are also known as osteochondromas. Additional sites of occurrence include on flat bones such as the pelvic bone and scapula. The distribution and number of these exostoses show a wide diversity among affected individuals. Exostoses usually present during childhood. The vast majority of affected individuals become clinically manifest by the time they reach adolescence. A small percentage of affected individuals are at risk for development of sarcomas as a result of malignant transformation. The incidence of hereditary multiple exostoses is around 1 in 50,000 individuals. Hereditary multiple osteochondromas is the preferred term used by the World Health Organization.

Chondrosarcoma is a bone sarcoma, a primary cancer composed of cells derived from transformed cells that produce cartilage. A chondrosarcoma is a member of a category of tumors of bone and soft tissue known as sarcomas. About 30% of bone sarcomas are chondrosarcomas. It is resistant to chemotherapy and radiotherapy. Unlike other primary bone sarcomas that mainly affect children and adolescents, a chondrosarcoma can present at any age. It more often affects the axial skeleton than the appendicular skeleton.

A benign tumor is a mass of cells (tumor) that does not invade neighboring tissue or metastasize. Compared to malignant (cancerous) tumors, benign tumors generally have a slower growth rate. Benign tumors have relatively well differentiated cells. They are often surrounded by an outer surface or stay contained within the epithelium. Common examples of benign tumors include moles and uterine fibroids.

Osteochondromas are the most common benign tumors of the bones. The tumors take the form of cartilage-capped bony projections or outgrowth on the surface of bones exostoses. It is characterized as a type of overgrowth that can occur in any bone where cartilage forms bone. Tumors most commonly affect long bones about the knee and in the forearm. Additionally, flat bones such as the pelvis and scapula may be affected. Hereditary multiple exostoses usually present during childhood. Yet, the vast majority of affected individuals become clinically manifest by the time they reach adolescence. Osteochondromas occur in 3% of the general population and represent 35% of all benign tumors and 8% of all bone tumors. The majority of these tumors are solitary non-hereditary lesions and approximately 15% of osteochondromas occur as hereditary multiple exostoses preferably known as hereditary multiple osteochondromas (HMOs). Osteochondromas do not result from injury and the exact cause remains unknown. Recent research has indicated that multiple osteochondromas is an autosomal dominant inherited disease. Germ line mutations in EXT1 and EXT2 genes located on chromosomes 8 and 11 have been associated with the cause of the disease. The treatment choice for osteochondroma is surgical removal of solitary lesion or partial excision of the outgrowth, when symptoms cause motion limitations or nerve and blood vessel impingements. In hereditary multiple exostoses the indications of surgery are based upon multiple factors that are taken collectively, namely: patient's age, tumor location and number, accompanying symptomatology, esthetic concerns, family history and underlying gene mutation. A variety of surgical procedures have been employed to remedy hereditary multiple exostoses such as osteochondroma excision, bone lengthening, corrective osteotomy and hemiepiphysiodesis. Sometimes a combination of the previous procedures is used. The indicators of surgical success in regard to disease and patient characteristics are greatly disputable. Because most studies of hereditary multiple exostoses are retrospective and of limited sample size with missing data, the best evidence for each of the currently practiced surgical procedures is lacking.

Myositis ossificans comprises two syndromes characterized by heterotopic ossification (calcification) of muscle. The World Health Organization, 2020, has grouped myositis ossificans together with fibro-osseous pseudotumor of digits as a single specific entity in the category of fibroblastic and myofibroblastic tumors.

Osteochondritis dissecans is a joint disorder primarily of the subchondral bone in which cracks form in the articular cartilage and the underlying subchondral bone. OCD usually causes pain during and after sports. In later stages of the disorder there will be swelling of the affected joint which catches and locks during movement. Physical examination in the early stages does only show pain as symptom, in later stages there could be an effusion, tenderness, and a crackling sound with joint movement.

Subungual exostosis is a type of non-cancerous bone tumor of the chondrogenic type, and consists of bone and cartilage. It usually projects from the upper surface of the big toe underlying the nailbed, giving rise to a painful swelling that destroys the nail. Subsequent ulceration and infection may occur.
Osteochondrodysplasia is a general term for a disorder of the development (dysplasia) of bone ("osteo") and cartilage ("chondro"). Osteochondrodysplasias are rare diseases. About 1 in 5,000 babies are born with some type of skeletal dysplasia. Nonetheless, if taken collectively, genetic skeletal dysplasias or osteochondrodysplasias comprise a recognizable group of genetically determined disorders with generalized skeletal affection. Osteochondrodysplasias can result in marked functional limitation and even mortality.

The epiphyseal plate is a hyaline cartilage plate in the metaphysis at each end of a long bone. It is the part of a long bone where new bone growth takes place; that is, the whole bone is alive, with maintenance remodeling throughout its existing bone tissue, but the growth plate is the place where the long bone grows longer.

Ollier disease is a rare sporadic nonhereditary skeletal disorder in which typically benign cartilaginous tumors (enchondromas) develop near the growth plate cartilage. This is caused by cartilage rests that grow and reside within the metaphysis or diaphysis and eventually mineralize over time to form multiple enchondromas. Key signs of the disorder include asymmetry and shortening of the limb as well as an increased thickness of the bone margin. These symptoms are typically first visible during early childhood with the mean age of diagnosis being 13 years of age. Many patients with Ollier disease are prone to develop other malignancies including bone sarcomas that necessitate treatment and the removal of malignant bone neoplasm. Cases in patients with Ollier disease has shown a link to IDH1, IDH2, and PTH1R gene mutations. Currently, there are no forms of treatment for the underlying condition of Ollier disease but complications such as fractures, deformities, malignancies that arise from it can be treated through surgical procedures. The prevalence of this condition is estimated at around 1 in 100,000. It is unclear whether the men or women are more affected by this disorder due to conflicting case studies.

Fairbank's disease or multiple epiphyseal dysplasia (MED) is a rare genetic disorder that affects the growing ends of bones. Long bones normally elongate by expansion of cartilage in the growth plate near their ends. As it expands outward from the growth plate, the cartilage mineralizes and hardens to become bone (ossification). In MED, this process is defective.

Pseudoachondroplasia is an inherited disorder of bone growth. It is a genetic autosomal dominant disorder. It is generally not discovered until 2–3 years of age, since growth is normal at first. Pseudoachondroplasia is usually first detected by a drop of linear growth in contrast to peers, a waddling gait or arising lower limb deformities.

An accessory bone or supernumerary bone is a bone that is not normally present in the body, but can be found as a variant in a significant number of people. It poses a risk of being misdiagnosed as bone fractures on radiography.
Fibrocartilaginous mesenchymoma of bone (FCMB) is an extremely rare tumor first described in 1984. About 26 cases have been reported in literature, with patient ages spanning from 9 to 25 years, though a case in a male infant aged 1 year and 7 months has been reported. Quick growth and bulky size are remarkable features of this tumor.

Orthopedic surgery is the branch of surgery concerned with conditions involving the musculoskeletal system. Orthopedic surgeons use both surgical and nonsurgical means to treat musculoskeletal injuries, sports injuries, degenerative diseases, infections, bone tumours, and congenital limb deformities. Trauma surgery and traumatology is a sub-specialty dealing with the operative management of fractures, major trauma and the multiply-injured patient.

Spondyloepimetaphyseal dysplasia-short limb-abnormal calcification syndrome is a rare genetic disorder which is characterized by osseous anomalies resulting in short stature and other afflictions.

