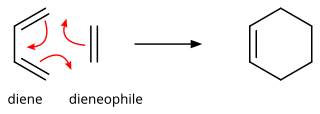
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels–Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.
An alkyne trimerisation is a [2+2+2] cycloaddition reaction in which three alkyne units react to form a benzene ring. The reaction requires a metal catalyst. The process is of historic interest as well as being applicable to organic synthesis. Being a cycloaddition reaction, it has high atom economy. Many variations have been developed, including cyclisation of mixtures of alkynes and alkenes as well as alkynes and nitriles.
In organic chemistry, the Knoevenagel condensation reaction is a type of chemical reaction named after German chemist Emil Knoevenagel. It is a modification of the aldol condensation.

The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl, driven by exergonically favored carbonyl CO bond formation Δ(ΔfH) = −327 kcal/mol (−1,370 kJ/mol).
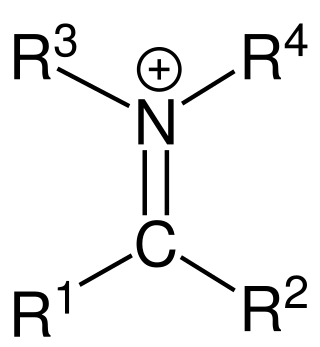
In organic chemistry, an iminium cation is a polyatomic ion with the general structure [R1R2C=NR3R4]+. They are common in synthetic chemistry and biology.

Wilhelm Rudolph Fittig was a German chemist. He discovered the pinacol coupling reaction, mesitylene, diacetyl and biphenyl. Fittig studied the action of sodium on ketones and hydrocarbons. He discovered the Fittig reaction or Wurtz–Fittig reaction for the synthesis of alkylbenzenes, he proposed a diketone structure for benzoquinone and isolated phenanthrene from coal tar. He discovered and synthesized the first lactones and investigated structures of piperine, naphthalene, and fluorene.
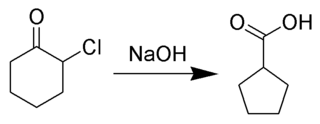
The Favorskii rearrangement is principally a rearrangement of cyclopropanones and α-halo ketones that leads to carboxylic acid derivatives. In the case of cyclic α-halo ketones, the Favorskii rearrangement constitutes a ring contraction. This rearrangement takes place in the presence of a base, sometimes hydroxide, to yield a carboxylic acid, but usually either an alkoxide base or an amine to yield an ester or an amide, respectively. α,α'-Dihaloketones eliminate HX under the reaction conditions to give α,β-unsaturated carbonyl compounds. Note that trihalomethyl ketone substrates will result in haloform and carboxylate formation via the haloform reaction instead.
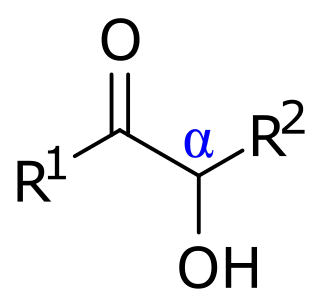
In organic chemistry, acyloins or α-hydroxy ketones are a class of organic compounds of the general form R−C(O)CH(OH)−R', composed of a hydroxy group adjacent to a ketone group. The name acyloin is derived from the fact that they are formally derived from reductive coupling of carboxylic acyl groups. They are one of the two main classes of hydroxy ketones, distinguished by the position of the hydroxy group relative to the ketone; in this form, the hydroxy is on the alpha carbon, explaining the secondary name of α-hydroxy ketone.
The Lossen rearrangement is the conversion of a hydroxamate ester to an isocyanate. Typically O-acyl, sulfonyl, or phosphoryl O-derivative are employed. The isocyanate can be used further to generate ureas in the presence of amines or generate amines in the presence of H2O.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
The Meerwein–Ponndorf–Verley (MPV) reduction in organic chemistry is the reduction of ketones and aldehydes to their corresponding alcohols utilizing aluminium alkoxide catalysis in the presence of a sacrificial alcohol. The advantages of the MPV reduction lie in its high chemoselectivity and its use of a cheap environmentally friendly metal catalyst. MPV reductions have been described as "obsolete" owing to the development of sodium borohydride and related reagents.
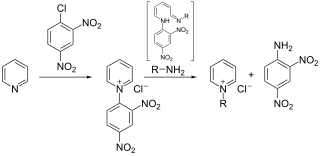
The Zincke reaction is an organic reaction, named after Theodor Zincke, in which a pyridine is transformed into a pyridinium salt by reaction with 2,4-dinitro-chlorobenzene and a primary amine.

In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds. Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
The Willgerodt rearrangement or Willgerodt reaction is an organic reaction converting an aryl alkyl ketone, alkyne, or alkene to the corresponding amide by reaction with ammonium polysulfide, named after Conrad Willgerodt. The formation of the corresponding carboxylic acid is a side reaction resulting from hydrolysis of the amide. When the alkyl group is an aliphatic chain, multiple reactions take place with the amide group always ending up at the terminal end. The net effect is thus migration of the carbonyl group to the end of the chain and oxidation.
The Debus–Radziszewski imidazole synthesis is a multi-component reaction used for the synthesis of imidazoles from a 1,2-dicarbonyl, an aldehyde, and ammonia or a primary amine. The method is used commercially to produce several imidazoles. The process is an example of a multicomponent reaction.

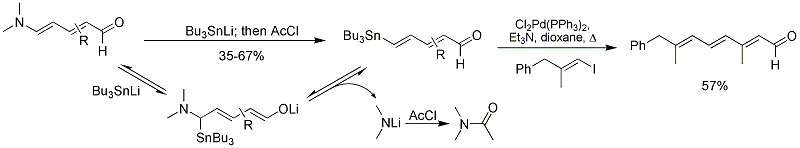
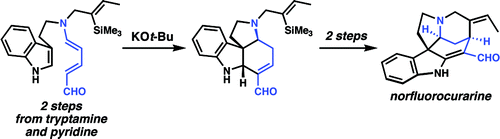
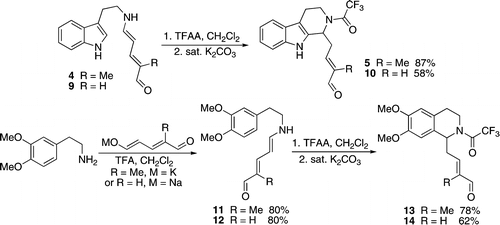
Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.
Rearrangements, especially those that can participate in cascade reactions, such as the aza-Cope rearrangements, are of high practical as well as conceptual importance in organic chemistry, due to their ability to quickly build structural complexity out of simple starting materials. The aza-Cope rearrangements are examples of heteroatom versions of the Cope rearrangement, which is a [3,3]-sigmatropic rearrangement that shifts single and double bonds between two allylic components. In accordance with the Woodward-Hoffman rules, thermal aza-Cope rearrangements proceed suprafacially. Aza-Cope rearrangements are generally classified by the position of the nitrogen in the molecule :
The asymmetric addition of alkynylzinc compounds to aldehydes is an example of a Nef synthesis, a chemical reaction whereby a chiral propargyl alcohol is prepared from a terminal alkyne and an aldehyde. This alkynylation reaction is enantioselective and involves an alkynylzinc reagent rather than the sodium acetylide used by John Ulric Nef in his 1899 report of the synthetic approach. Propargyl alcohols are versatile precursors for the chirally-selective synthesis of natural products and pharmaceutical agents, making this asymmetric addition reaction of alkynylzinc compounds useful. For example, Erick Carreira used this approach in a total synthesis of the marine natural product leucascandrolide A, a bioactive metabolite of the calcareous sponge Leucascandra caveolata with cytotoxic and antifungal properties isolated in 1996.

2-Carboxybenzaldehyde is a chemical compound. It consists of a benzene ring, with an aldehyde and a carboxylic acid as substituents that are ortho to each other. The compound exhibits ring–chain tautomerism: the two substituents can react with each other to form 3-hydroxyphthalide, a cyclic lactol. This lactol reacts readily with Grignard reagents, forming alkyl- and aryl-substituted phthalides. Other benzo-fused heterocyclic compounds can be derived from 2-carboxybenzaldehyde, including isoindolinones and phthalazinones, with a variety of pharmacological properties, such as the antihistamine azelastine.