
Motor neuron diseases or motor neurone diseases (MNDs) are a group of rare neurodegenerative disorders that selectively affect motor neurons, the cells which control voluntary muscles of the body. They include amyotrophic lateral sclerosis (ALS), progressive bulbar palsy (PBP), pseudobulbar palsy, progressive muscular atrophy (PMA), primary lateral sclerosis (PLS), spinal muscular atrophy (SMA) and monomelic amyotrophy (MMA), as well as some rarer variants resembling ALS.
Neuromyotonia (NMT) is a form of peripheral nerve hyperexcitability that causes spontaneous muscular activity resulting from repetitive motor unit action potentials of peripheral origin. NMT along with Morvan's syndrome are the most severe types in the Peripheral Nerve Hyperexciteability spectrum. Example of two more common and less severe syndromes in the spectrum are cramp fasciculation syndrome and benign fasciculation syndrome. NMT can have both hereditary and acquired (non-inherited) forms. The prevalence of NMT is unknown.

Transverse myelitis (TM) is a rare neurological condition wherein the spinal cord is inflamed. The adjective transverse implies that the spinal inflammation (myelitis) extends horizontally throughout the cross section of the spinal cord; the terms partial transverse myelitis and partial myelitis are sometimes used to specify inflammation that affects only part of the width of the spinal cord. TM is characterized by weakness and numbness of the limbs, deficits in sensation and motor skills, dysfunctional urethral and anal sphincter activities, and dysfunction of the autonomic nervous system that can lead to episodes of high blood pressure. Signs and symptoms vary according to the affected level of the spinal cord. The underlying cause of TM is unknown. The spinal cord inflammation seen in TM has been associated with various infections, immune system disorders, or damage to nerve fibers, by loss of myelin. As opposed to leukomyelitis which affects only the white matter, it affects the entire cross-section of the spinal cord. Decreased electrical conductivity in the nervous system can result.

Benign fasciculation syndrome (BFS) is characterized by fasciculation (twitching) of voluntary muscles in the body. The twitching can occur in any voluntary muscle group but is most common in the eyelids, arms, hands, fingers, legs, and feet. The tongue can also be affected. The twitching may be occasional to continuous. BFS must be distinguished from other conditions that include muscle twitches.

Onuf's nucleus is a distinct group of neurons located in the ventral part of the anterior horn of the sacral region of the human spinal cord involved in the maintenance of micturition and defecatory continence, as well as muscular contraction during orgasm. It contains motor neurons, and is the origin of the pudendal nerve. The sacral region of the spinal cord is the fourth segment of vertebrae in the spinal cord which consists of the vertebrae 26-30. While working in New York City in 1899, Bronislaw Onuf-Onufrowicz discovered this group of unique cells and originally identified it as “Group X.” “Group X” was considered distinct by Onufrowicz because the cells were different in size from the surrounding neurons in the anterolateral group, suggesting that they were independent.

Cervical spine disorders are illnesses that affect the cervical spine, which is made up of the upper first seven vertebrae, encasing and shielding the spinal cord. This fragment of the spine starts from the region above the shoulder blades and ends by supporting and connecting the skull.
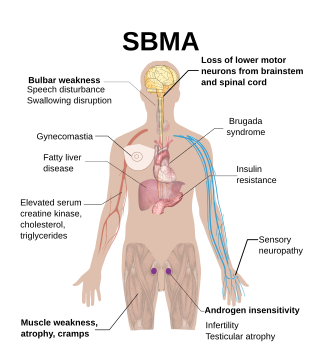
Spinal and bulbar muscular atrophy (SBMA), popularly known as Kennedy's disease, is a rare, adult-onset, X-linked recessive lower motor neuron disease caused by trinucleotide CAG repeat expansions in exon 1 of the androgen receptor (AR) gene, which results in both loss of AR function and toxic gain of function.
Primary lateral sclerosis (PLS) is a very rare neuromuscular disease characterized by progressive muscle weakness in the voluntary muscles. PLS belongs to a group of disorders known as motor neuron diseases. Motor neuron diseases develop when the nerve cells that control voluntary muscle movement degenerate and die, causing weakness in the muscles they control.
Beevor's sign is medical sign in which the navel moves towards the head upon flexing the neck, indicating selective weakness of the lower abdominal muscles. Causes include spinal cord injury, amyotrophic lateral sclerosis (ALS), and facioscapulohumeral muscular dystrophy (FSHD).

A neuromuscular disease is any disease affecting the peripheral nervous system (PNS), the neuromuscular junctions, or skeletal muscles, all of which are components of the motor unit. Damage to any of these structures can cause muscle atrophy and weakness. Issues with sensation can also occur.

Progressive muscular atrophy (PMA), also called Duchenne–Aran disease and Duchenne–Aran muscular atrophy, is a disorder characterised by the degeneration of lower motor neurons, resulting in generalised, progressive loss of muscle function.
Progressive bulbar palsy (PBP) is a medical condition. It belongs to a group of disorders known as motor neuron diseases. PBP is a disease that attacks the nerves supplying the bulbar muscles. These disorders are characterized by the degeneration of motor neurons in the cerebral cortex, spinal cord, brain stem, and pyramidal tracts. This specifically involves the glossopharyngeal nerve (IX), vagus nerve (X), and hypoglossal nerve (XII).
In medicine, split hand syndrome is a neurological syndrome in which the hand muscles on the side of the thumb appear wasted, whereas the muscles on the side of the little finger are spared. Anatomically, the abductor pollicis brevis and first dorsal interosseous muscle are more wasted than the abductor digiti minimi.
Madras motor neuron disease is a rare motor neuron disease originating in South India. Two other forms of the disease have been found, Familial Madras Motor Neuron Disease (FMMND) and the variant Madras Motor Neuron Disease (MMNDV). The symptoms of MMND include weakness in the arms and legs, loss of vision, and deafness. Most affected individuals are diagnosed by the age of 15 and occurs at the same frequency in males and females. While the cause of the disease and its origins are not yet known, supportive care is available to individuals affected by the disease.

Amyotrophic lateral sclerosis (ALS), also known as motor neurone disease (MND) or Lou Gehrig's disease in the United States, is a rare but terminal neurodegenerative disorder that results in the progressive loss of both upper and lower motor neurons that normally control voluntary muscle contraction. ALS is the most common form of the motor neuron diseases. ALS often presents in its early stages with gradual muscle stiffness, twitches, weakness, and wasting. Motor neuron loss typically continues until the abilities to eat, speak, move, and, lastly, breathe are all lost. While only 15% of people with ALS also fully develop frontotemporal dementia, an estimated 50% face at least some minor difficulties with thinking and behavior. Depending on which of the aforementioned symptoms develops first, ALS is classified as limb-onset or bulbar-onset.

X-linked spinal muscular atrophy type 2, also known as arthrogryposis multiplex congenita X-linked type 1 (AMCX1), is a rare neurological disorder involving death of motor neurons in the anterior horn of spinal cord resulting in generalised muscle wasting (atrophy). The disease is caused by a mutation in UBA1 gene and is passed in an X-linked recessive manner by carrier mothers to affected sons.
Cramp fasciculation syndrome (CFS) is a rare peripheral nerve hyperexcitability disorder. It is more severe than the related disorder known as benign fasciculation syndrome; it causes fasciculations, cramps, pain, fatigue, and muscle stiffness similar to those seen in neuromyotonia. Patients with CFS, like those with neuromyotonia, may also experience paresthesias. Most cases of cramp fasciculation syndrome are idiopathic.

Spinal stenosis is an abnormal narrowing of the spinal canal or neural foramen that results in pressure on the spinal cord or nerve roots. Symptoms may include pain, numbness, or weakness in the arms or legs. Symptoms are typically gradual in onset and improve with leaning forward. Severe symptoms may include loss of bladder control, loss of bowel control, or sexual dysfunction.
Sunil Pradhan is an Indian neurologist, medical researcher and writer, known for the invention of two electrophysiological techniques. He has also described five medical signs, of which one related to Duchenne muscular dystrophy is known as Pradhan Sign, and the others associated with facioscapulohumeral muscular dystrophy (FSHD) and similar neuro diseases. The Government of India awarded him the Padma Shri, the fourth highest civilian award, in 2014 for his contributions to the field of neuroscience.

