
Eosinophilia is a condition in which the eosinophil count in the peripheral blood exceeds 5×108/L (500/μL). Hypereosinophilia is an elevation in an individual's circulating blood eosinophil count above 1.5 × 109/L (i.e. 1,500/μL). The hypereosinophilic syndrome is a sustained elevation in this count above 1.5 × 109/L (i.e. 1,500/μL) that is also associated with evidence of eosinophil-based tissue injury.

Familial Mediterranean fever (FMF) is a hereditary inflammatory disorder. FMF is an autoinflammatory disease caused by mutations in Mediterranean fever gene, which encodes a 781–amino acid protein called pyrin. While all ethnic groups are susceptible to FMF, it usually occurs in people of Mediterranean origin—including Sephardic Jews, Mizrahi Jews, Ashkenazi Jews, Assyrians, Armenians, Azerbaijanis, Druze, Levantines, Kurds, Greeks, Turks and Italians.

Hives, also known as urticaria, is a kind of skin rash with red, raised, itchy bumps. Hives may burn or sting. The patches of rash may appear on different body parts, with variable duration from minutes to days, and does not leave any long-lasting skin change. Fewer than 5% of cases last for more than six weeks. The condition frequently recurs.
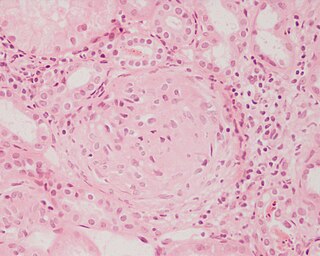
Glomerulonephritis (GN) is a term used to refer to several kidney diseases. Many of the diseases are characterised by inflammation either of the glomeruli or of the small blood vessels in the kidneys, hence the name, but not all diseases necessarily have an inflammatory component.

Cold urticaria is a disorder in which large red welts called hives (urticaria) form on the skin after exposure to a cold stimulus. The hives are usually itchy and often the hands, feet and other parts of the body will become itchy and swollen as well. Hives vary in size from about 7 mm in diameter to as big as about 27 mm or larger.
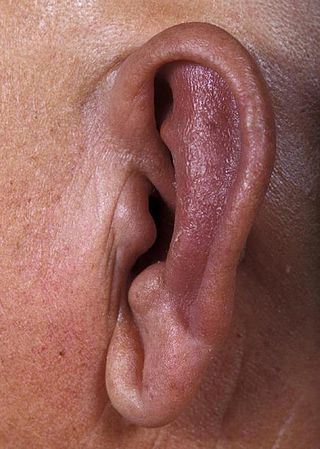
Relapsing polychondritis is a systemic disease characterized by repeated episodes of inflammation and in some cases deterioration of cartilage. The disease can be life-threatening if the respiratory tract, heart valves, or blood vessels are affected. The exact mechanism is poorly understood.
Fever of unknown origin (FUO) refers to a condition in which the patient has an elevated temperature (fever) but, despite investigations by one or more qualified physicians, no explanation is found.

Muckle–Wells syndrome (MWS) is a rare autosomal dominant disease which causes sensorineural deafness and recurrent hives, and can lead to amyloidosis. Individuals with MWS often have episodic fever, chills, and joint pain. As a result, MWS is considered a type of periodic fever syndrome. MWS is caused by a defect in the CIAS1 gene which creates the protein cryopyrin. MWS is closely related to two other syndromes, familial cold urticaria and neonatal onset multisystem inflammatory disease—in fact, all three are related to mutations in the same gene and subsumed under the term cryopyrin-associated periodic syndromes (CAPS).
Periodic fever syndromes are a set of disorders characterized by recurrent episodes of systemic and organ-specific inflammation. Unlike autoimmune disorders such as systemic lupus erythematosus, in which the disease is caused by abnormalities of the adaptive immune system, people with autoinflammatory diseases do not produce autoantibodies or antigen-specific T or B cells. Instead, the autoinflammatory diseases are characterized by errors in the innate immune system.

NLR family pyrin domain containing 3 (NLRP3), is a protein that in humans is encoded by the NLRP3 gene located on the long arm of chromosome 1.
Chronic recurrent multifocal osteomyelitis (CRMO) is a rare condition (1:1,000,000), in which the bones have lesions, inflammation, and pain. It is called multifocal because it can appear in different parts of the body, primarily bones, and osteomyelitis because it is very similar to that disease, although CRMO appears to be without any infection.
Anetoderma is a benign but uncommon disorder that causes localized areas of flaccid or herniated sac-like skin due to a focal reduction of dermal elastic tissue. Anetoderma is subclassified as primary anetoderma, secondary anetoderma, iatrogenic anetoderma of prematurity, congenital anetoderma, familial anetoderma, and drug-induced anetoderma.

Blau syndrome is an autosomal dominant genetic inflammatory disorder which affects the skin, eyes, and joints. It is caused by a mutation in the NOD2 (CARD15) gene. and is classified as an inborn errors of immunity. Symptoms usually begin before the age of four, and the disease manifests as early onset cutaneous sarcoidosis, granulomatous arthritis, and uveitis.
PAPA syndrome is a rare genetic disorder characterised by its effects on skin and joints. The acronym PAPA stands for pyogenic arthritis, pyoderma gangrenosum and acne.
Cryopyrin-associated periodic syndrome (CAPS) is a group of rare, heterogeneous autoinflammatory disease characterized by interleukin 1β-mediated systemic inflammation and clinical symptoms involving skin, joints, central nervous system, and eyes. It encompasses a spectrum of three clinically overlapping autoinflammatory syndromes including familial cold autoinflammatory syndrome, the Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease that were originally thought to be distinct entities, but in fact share a single genetic mutation and pathogenic pathway, and keratoendotheliitis fugax hereditaria in which the autoinflammatory symptoms affect only the anterior segment of the eye.

Behçet's disease (BD) is a type of inflammatory disorder which affects multiple parts of the body. The most common symptoms include painful sores on the mucous membranes of the mouth and other parts of the body, inflammation of parts of the eye, and arthritis. The sores can last from a few days, up to a week or more. Less commonly there may be inflammation of the brain or spinal cord, blood clots, aneurysms, or blindness. Often, the symptoms come and go.

Aicardi–Goutières syndrome (AGS), which is completely distinct from the similarly named Aicardi syndrome, is a rare, usually early onset childhood, inflammatory disorder most typically affecting the brain and the skin. The majority of affected individuals experience significant intellectual and physical problems, although this is not always the case. The clinical features of AGS can mimic those of in utero acquired infection, and some characteristics of the condition also overlap with the autoimmune disease systemic lupus erythematosus (SLE). Following an original description of eight cases in 1984, the condition was first referred to as 'Aicardi–Goutières syndrome' (AGS) in 1992, and the first international meeting on AGS was held in Pavia, Italy, in 2001.
NSAIDhypersensitivity reactions encompass a broad range of allergic or allergic-like symptoms that occur within minutes to hours after ingesting aspirin or other NSAID nonsteroidal anti-inflammatory drugs. Hypersensitivity drug reactions differ from drug toxicity reactions in that drug toxicity reactions result from the pharmacological action of a drug, are dose-related, and can occur in any treated individual. Hypersensitivity reactions are idiosyncratic reactions to a drug. Although the term NSAID was introduced to signal a comparatively low risk of adverse effects, NSAIDs do evoke a broad range of hypersensitivity syndromes. These syndromes have recently been classified by the European Academy of Allergy and Clinical Immunology Task Force on NSAIDs Hypersensitivity.
Autoinflammatory diseases (AIDs) are a group of rare disorders caused by dysfunction of the innate immune system. These responses are characterized by periodic or chronic systemic inflammation, usually without the involvement of adaptive immunity.
