
In biology, histones are highly basic proteins abundant in lysine and arginine residues that are found in eukaryotic cell nuclei and in most Archaeal phyla. They act as spools around which DNA winds to create structural units called nucleosomes. Nucleosomes in turn are wrapped into 30-nanometer fibers that form tightly packed chromatin. Histones prevent DNA from becoming tangled and protect it from DNA damage. In addition, histones play important roles in gene regulation and DNA replication. Without histones, unwound DNA in chromosomes would be very long. For example, each human cell has about 1.8 meters of DNA if completely stretched out; however, when wound about histones, this length is reduced to about 90 micrometers (0.09 mm) of 30 nm diameter chromatin fibers.
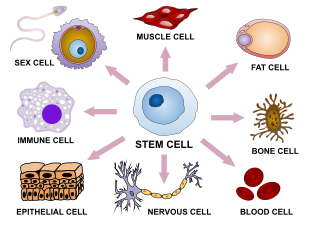
Cellular differentiation is the process in which a stem cell changes from one type to a differentiated one. Usually, the cell changes to a more specialized type. Differentiation happens multiple times during the development of a multicellular organism as it changes from a simple zygote to a complex system of tissues and cell types. Differentiation continues in adulthood as adult stem cells divide and create fully differentiated daughter cells during tissue repair and during normal cell turnover. Some differentiation occurs in response to antigen exposure. Differentiation dramatically changes a cell's size, shape, membrane potential, metabolic activity, and responsiveness to signals. These changes are largely due to highly controlled modifications in gene expression and are the study of epigenetics. With a few exceptions, cellular differentiation almost never involves a change in the DNA sequence itself. However, metabolic composition does get altered quite dramatically where stem cells are characterized by abundant metabolites with highly unsaturated structures whose levels decrease upon differentiation. Thus, different cells can have very different physical characteristics despite having the same genome.

Histone H4 is one of the five main histone proteins involved in the structure of chromatin in eukaryotic cells. Featuring a main globular domain and a long N-terminal tail, H4 is involved with the structure of the nucleosome of the 'beads on a string' organization. Histone proteins are highly post-translationally modified. Covalently bonded modifications include acetylation and methylation of the N-terminal tails. These modifications may alter expression of genes located on DNA associated with its parent histone octamer. Histone H4 is an important protein in the structure and function of chromatin, where its sequence variants and variable modification states are thought to play a role in the dynamic and long term regulation of genes.
Histone methylation is a process by which methyl groups are transferred to amino acids of histone proteins that make up nucleosomes, which the DNA double helix wraps around to form chromosomes. Methylation of histones can either increase or decrease transcription of genes, depending on which amino acids in the histones are methylated, and how many methyl groups are attached. Methylation events that weaken chemical attractions between histone tails and DNA increase transcription because they enable the DNA to uncoil from nucleosomes so that transcription factor proteins and RNA polymerase can access the DNA. This process is critical for the regulation of gene expression that allows different cells to express different genes.

Histone-modifying enzymes are enzymes involved in the modification of histone substrates after protein translation and affect cellular processes including gene expression. To safely store the eukaryotic genome, DNA is wrapped around four core histone proteins, which then join to form nucleosomes. These nucleosomes further fold together into highly condensed chromatin, which renders the organism's genetic material far less accessible to the factors required for gene transcription, DNA replication, recombination and repair. Subsequently, eukaryotic organisms have developed intricate mechanisms to overcome this repressive barrier imposed by the chromatin through histone modification, a type of post-translational modification which typically involves covalently attaching certain groups to histone residues. Once added to the histone, these groups elicit either a loose and open histone conformation, euchromatin, or a tight and closed histone conformation, heterochromatin. Euchromatin marks active transcription and gene expression, as the light packing of histones in this way allows entry for proteins involved in the transcription process. As such, the tightly packed heterochromatin marks the absence of current gene expression.
Chromatin remodeling is the dynamic modification of chromatin architecture to allow access of condensed genomic DNA to the regulatory transcription machinery proteins, and thereby control gene expression. Such remodeling is principally carried out by 1) covalent histone modifications by specific enzymes, e.g., histone acetyltransferases (HATs), deacetylases, methyltransferases, and kinases, and 2) ATP-dependent chromatin remodeling complexes which either move, eject or restructure nucleosomes. Besides actively regulating gene expression, dynamic remodeling of chromatin imparts an epigenetic regulatory role in several key biological processes, egg cells DNA replication and repair; apoptosis; chromosome segregation as well as development and pluripotency. Aberrations in chromatin remodeling proteins are found to be associated with human diseases, including cancer. Targeting chromatin remodeling pathways is currently evolving as a major therapeutic strategy in the treatment of several cancers.
An epigenetic clock is a biochemical test that can be used to measure age. The test is based on DNA methylation levels, measuring the accumulation of methyl groups to one's DNA molecules.
H3K4me3 is an epigenetic modification to the DNA packaging protein Histone H3 that indicates tri-methylation at the 4th lysine residue of the histone H3 protein and is often involved in the regulation of gene expression. The name denotes the addition of three methyl groups (trimethylation) to the lysine 4 on the histone H3 protein.
H3K27me3 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the tri-methylation of lysine 27 on histone H3 protein.
H3K9me3 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the tri-methylation at the 9th lysine residue of the histone H3 protein and is often associated with heterochromatin.
H3K9me2 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the di-methylation at the 9th lysine residue of the histone H3 protein. H3K9me2 is strongly associated with transcriptional repression. H3K9me2 levels are higher at silent compared to active genes in a 10kb region surrounding the transcriptional start site. H3K9me2 represses gene expression both passively, by prohibiting acetylation as therefore binding of RNA polymerase or its regulatory factors, and actively, by recruiting transcriptional repressors. H3K9me2 has also been found in megabase blocks, termed Large Organised Chromatin K9 domains (LOCKS), which are primarily located within gene-sparse regions but also encompass genic and intergenic intervals. Its synthesis is catalyzed by G9a, G9a-like protein, and PRDM2. H3K9me2 can be removed by a wide range of histone lysine demethylases (KDMs) including KDM1, KDM3, KDM4 and KDM7 family members. H3K9me2 is important for various biological processes including cell lineage commitment, the reprogramming of somatic cells to induced pluripotent stem cells, regulation of the inflammatory response, and addiction to drug use.
H3K4me1 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the mono-methylation at the 4th lysine residue of the histone H3 protein and often associated with gene enhancers.
H3K36me3 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the tri-methylation at the 36th lysine residue of the histone H3 protein and often associated with gene bodies.
H4K20me is an epigenetic modification to the DNA packaging protein Histone H4. It is a mark that indicates the mono-methylation at the 20th lysine residue of the histone H4 protein. This mark can be di- and tri-methylated. It is critical for genome integrity including DNA damage repair, DNA replication and chromatin compaction.
H4K16ac is an epigenetic modification to the DNA packaging protein Histone H4. It is a mark that indicates the acetylation at the 16th lysine residue of the histone H4 protein.
H3K36me2 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the di-methylation at the 36th lysine residue of the histone H3 protein.
H3K36me is an epigenetic modification to the DNA packaging protein Histone H3, specifically, the mono-methylation at the 36th lysine residue of the histone H3 protein.
H3R42me is an epigenetic modification to the DNA packaging protein histone H3. It is a mark that indicates the mono-methylation at the 42nd arginine residue of the histone H3 protein. In epigenetics, arginine methylation of histones H3 and H4 is associated with a more accessible chromatin structure and thus higher levels of transcription. The existence of arginine demethylases that could reverse arginine methylation is controversial.
H3R2me2 is an epigenetic modification to the DNA packaging protein histone H3. It is a mark that indicates the di-methylation at the 2nd arginine residue of the histone H3 protein. In epigenetics, arginine methylation of histones H3 and H4 is associated with a more accessible chromatin structure and thus higher levels of transcription. The existence of arginine demethylases that could reverse arginine methylation is controversial.
H3T11P is an epigenetic modification to the DNA packaging protein histone H3. It is a mark that indicates the phosphorylation the 11th threonine residue of the histone H3 protein.
