Myoclonus is a brief, involuntary, irregular twitching of a muscle, a joint, or a group of muscles, different from clonus, which is rhythmic or regular. Myoclonus describes a medical sign and, generally, is not a diagnosis of a disease. It belongs to the hyperkinetic movement disorders, among tremor and chorea for example. These myoclonic twitches, jerks, or seizures are usually caused by sudden muscle contractions or brief lapses of contraction. The most common circumstance under which they occur is while falling asleep. Myoclonic jerks occur in healthy people and are experienced occasionally by everyone. However, when they appear with more persistence and become more widespread they can be a sign of various neurological disorders. Hiccups are a kind of myoclonic jerk specifically affecting the diaphragm. When a spasm is caused by another person it is known as a provoked spasm. Shuddering attacks in babies fall in this category.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.
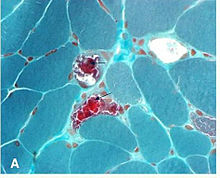
Kearns–Sayre syndrome (KSS), oculocraniosomatic disorder or oculocranionsomatic neuromuscular disorder with ragged red fibers is a mitochondrial myopathy with a typical onset before 20 years of age. KSS is a more severe syndromic variant of chronic progressive external ophthalmoplegia, a syndrome that is characterized by isolated involvement of the muscles controlling movement of the eyelid and eye. This results in ptosis and ophthalmoplegia respectively. KSS involves a combination of the already described CPEO as well as pigmentary retinopathy in both eyes and cardiac conduction abnormalities. Other symptoms may include cerebellar ataxia, proximal muscle weakness, deafness, diabetes mellitus, growth hormone deficiency, hypoparathyroidism, and other endocrinopathies. In both of these diseases, muscle involvement may begin unilaterally but always develops into a bilateral deficit, and the course is progressive. This discussion is limited specifically to the more severe and systemically involved variant.

MELAS is one of the family of mitochondrial diseases, which also include MIDD, MERRF syndrome, and Leber's hereditary optic neuropathy. It was first characterized under this name in 1984. A feature of these diseases is that they are caused by defects in the mitochondrial genome which is inherited purely from the female parent. The most common MELAS mutation is mitochondrial mutation, mtDNA, referred to as m.3243A>G.
Myoclonic epilepsy refers to a family of epilepsies that present with myoclonus. When myoclonic jerks are occasionally associated with abnormal brain wave activity, it can be categorized as myoclonic seizure. If the abnormal brain wave activity is persistent and results from ongoing seizures, then a diagnosis of myoclonic epilepsy may be considered.
Mitochondrial neurogastrointestinal encephalopathy syndrome (MNGIE) is a rare autosomal recessive mitochondrial disease. It has been previously referred to as polyneuropathy, ophthalmoplegia, leukoencephalopathy, and intestinal pseudoobstruction. The disease presents in childhood, but often goes unnoticed for decades. Unlike typical mitochondrial diseases caused by mitochondrial DNA (mtDNA) mutations, MNGIE is caused by mutations in the TYMP gene, which encodes the enzyme thymidine phosphorylase. Mutations in this gene result in impaired mitochondrial function, leading to intestinal symptoms as well as neuro-ophthalmologic abnormalities. A secondary form of MNGIE, called MNGIE without leukoencephalopathy, can be caused by mutations in the POLG gene.
Progressive Myoclonic Epilepsies (PME) are a rare group of inherited neurodegenerative diseases characterized by myoclonus, resistance to treatment, and neurological deterioration. The cause of PME depends largely on the type of PME. Most PMEs are caused by autosomal dominant or recessive and mitochondrial mutations. The location of the mutation also affects the inheritance and treatment of PME. Diagnosing PME is difficult due to their genetic heterogeneity and the lack of a genetic mutation identified in some patients. The prognosis depends largely on the worsening symptoms and failure to respond to treatment. There is no current cure for PME and treatment focuses on managing myoclonus and seizures through antiepileptic medication (AED).
Chronic progressive external ophthalmoplegia (CPEO) is a type of eye disorder characterized by slowly progressive inability to move the eyes and eyebrows. It is often the only feature of mitochondrial disease, in which case the term CPEO may be given as the diagnosis. In other people suffering from mitochondrial disease, CPEO occurs as part of a syndrome involving more than one part of the body, such as Kearns–Sayre syndrome. Occasionally CPEO may be caused by conditions other than mitochondrial diseases.
Mitochondrially encoded tRNA leucine 1 (UUA/G) also known as MT-TL1 is a transfer RNA which in humans is encoded by the mitochondrial MT-TL1 gene.
Mitochondrially encoded tRNA histidine, also known as MT-TH, is a transfer RNA which, in humans, is encoded by the mitochondrial MT-TH gene.
Mitochondrially encoded tRNA aspartic acid also known as MT-TD is a transfer RNA which in humans is encoded by the mitochondrial MT-TD gene.
Mitochondrially encoded tRNA glutamic acid also known as MT-TE is a transfer RNA which in humans is encoded by the mitochondrial MT-TE gene. MT-TE is a small 69 nucleotide RNA that transfers the amino acid glutamic acid to a growing polypeptide chain at the ribosome site of protein synthesis during translation.
Mitochondrially encoded tRNA phenylalanine also known as MT-TF is a transfer RNA which in humans is encoded by the mitochondrial MT-TF gene.
Mitochondrially encoded tRNA glycine also known as MT-TG is a transfer RNA which in humans is encoded by the mitochondrial MT-TG gene.
Mitochondrially encoded tRNA isoleucine also known as MT-TI is a transfer RNA which in humans is encoded by the mitochondrial MT-TI gene.
Mitochondrially encoded tRNA lysine also known as MT-TK is a transfer RNA which in humans is encoded by the mitochondrial MT-TK gene.
Mitochondrially encoded tRNA leucine 2 (CUN) also known as MT-TL2 is a transfer RNA which in humans is encoded by the mitochondrial MT-TL2 gene.
Mitochondrially encoded tRNA proline also known as MT-TP is a transfer RNA that in humans is encoded by the mitochondrial MT-TP gene.
Mitochondrially encoded tRNA threonine also known as MT-TT is a transfer RNA which in humans is encoded by the mitochondrial MT-TT gene.
Mitochondrially encoded tRNA tyrosine, also known as MT-TY, is a transfer RNA which in humans is encoded by the mitochondrial MT-TY gene.