An axon, or nerve fiber, is a long, slender projection of a nerve cell, or neuron, in vertebrates, that typically conducts electrical impulses known as action potentials away from the nerve cell body. The function of the axon is to transmit information to different neurons, muscles, and glands. In certain sensory neurons, such as those for touch and warmth, the axons are called afferent nerve fibers and the electrical impulse travels along these from the periphery to the cell body and from the cell body to the spinal cord along another branch of the same axon. Axon dysfunction can be the cause of many inherited and acquired neurological disorders that affect both the peripheral and central neurons. Nerve fibers are classed into three types – group A nerve fibers, group B nerve fibers, and group C nerve fibers. Groups A and B are myelinated, and group C are unmyelinated. These groups include both sensory fibers and motor fibers. Another classification groups only the sensory fibers as Type I, Type II, Type III, and Type IV.

Myelin is a lipid-rich extramembranous structure found on the axons and dendrites of neuron in many bilaterian animals, mainly vertebrates, as well as some arthropods and annelids. A circumferential wrapping of myelin, known as a myelin sheath, increases the conduction speed of electrical impulses passing along the axon by generating saltatory conductions, which are much faster than conduction along an unmyelinated axon, and also reduce signal loss due to extrinsic disturbances.
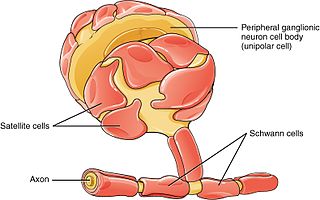
Schwann cells or neurolemmocytes are the principal glia of the peripheral nervous system (PNS). Glial cells function to support neurons and in the PNS, also include satellite cells, olfactory ensheathing cells, enteric glia and glia that reside at sensory nerve endings, such as the Pacinian corpuscle. The two types of Schwann cells are myelinating and nonmyelinating. Myelinating Schwann cells wrap around axons of motor and sensory neurons to form the myelin sheath. The Schwann cell promoter is present in the downstream region of the human dystrophin gene that gives shortened transcript that are again synthesized in a tissue-specific manner.
A motor nerve is a nerve that transmits motor signals from the central nervous system (CNS) to the muscles of the body. This is different from the motor neuron, which includes a cell body and branching of dendrites, while the nerve is made up of a bundle of axons. Motor nerves act as efferent nerves which carry information out from the CNS to muscles, as opposed to afferent nerves, which transfer signals from sensory receptors in the periphery to the CNS. Efferent nerves can also connect to glands or other organs/issues instead of muscles. In addition, there are nerves that serve as both sensory and motor nerves called mixed nerves.

Oligodendrocytes, also known as oligodendroglia, are a type of neuroglia whose main functions are to provide support and insulation to axons within the central nervous system (CNS) of jawed vertebrates. Their function is similar to that of Schwann cells, which perform the same task in the peripheral nervous system (PNS). Oligodendrocytes accomplish this by forming the myelin sheath around axons. Unlike Schwann cells, a single oligodendrocyte can extend its processes to cover around 50 axons, with each axon being wrapped in approximately 1 μm of myelin sheath. Furthermore, an oligodendrocyte can provide myelin segments for multiple adjacent axons.

Glia, also called glial cells(gliocytes) or neuroglia, are non-neuronal cells in the central nervous system (brain and spinal cord) and the peripheral nervous system that do not produce electrical impulses. The neuroglia make up more than one half the volume of neural tissue in our body. They maintain homeostasis, form myelin in the peripheral nervous system, and provide support and protection for neurons. In the central nervous system, glial cells include oligodendrocytes, astrocytes, ependymal cells and microglia, and in the peripheral nervous system they include Schwann cells and satellite cells.
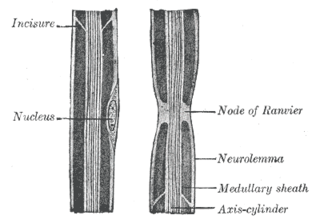
In neuroscience and anatomy, nodes of Ranvier, also known as myelin-sheath gaps, occur along a myelinated axon where the axolemma is exposed to the extracellular space. Nodes of Ranvier are uninsulated and highly enriched in ion channels, allowing them to participate in the exchange of ions required to regenerate the action potential. Nerve conduction in myelinated axons is referred to as saltatory conduction due to the manner in which the action potential seems to "jump" from one node to the next along the axon. This results in faster conduction of the action potential.

Astrogliosis is an abnormal increase in the number of astrocytes due to the destruction of nearby neurons from central nervous system (CNS) trauma, infection, ischemia, stroke, autoimmune responses or neurodegenerative disease. In healthy neural tissue, astrocytes play critical roles in energy provision, regulation of blood flow, homeostasis of extracellular fluid, homeostasis of ions and transmitters, regulation of synapse function and synaptic remodeling. Astrogliosis changes the molecular expression and morphology of astrocytes, in response to infection for example, in severe cases causing glial scar formation that may inhibit axon regeneration.
Axonotmesis is an injury to the peripheral nerve of one of the extremities of the body. The axons and their myelin sheath are damaged in this kind of injury, but the endoneurium, perineurium and epineurium remain intact. Motor and sensory functions distal to the point of injury are completely lost over time leading to Wallerian degeneration due to ischemia, or loss of blood supply. Axonotmesis is usually the result of a more severe crush or contusion than neurapraxia.
Gliosis is a nonspecific reactive change of glial cells in response to damage to the central nervous system (CNS). In most cases, gliosis involves the proliferation or hypertrophy of several different types of glial cells, including astrocytes, microglia, and oligodendrocytes. In its most extreme form, the proliferation associated with gliosis leads to the formation of a glial scar.
Remyelination is the process of propagating oligodendrocyte precursor cells to form oligodendrocytes to create new myelin sheaths on demyelinated axons in the CNS. This is a process naturally regulated in the body and tends to be very efficient in a healthy CNS. The process creates a thinner myelin sheath than normal, but it helps to protect the axon from further damage, from overall degeneration, and proves to increase conductance once again. The processes underlying remyelination are under investigation in the hope of finding treatments for demyelinating diseases, such as multiple sclerosis.

Myelin-associated glycoprotein is a type 1 transmembrane protein glycoprotein localized in periaxonal Schwann cell and oligodendrocyte membranes, where it plays a role in glial-axonal interactions. MAG is a member of the SIGLEC family of proteins and is a functional ligand of the NOGO-66 receptor, NgR. MAG is believed to be involved in myelination during nerve regeneration in the PNS and is vital for the long-term survival of the myelinated axons following myelinogenesis. In the CNS MAG is one of three main myelin-associated inhibitors of axonal regeneration after injury, making it an important protein for future research on neurogenesis in the CNS.

Nerve injury is an injury to nervous tissue. There is no single classification system that can describe all the many variations of nerve injuries. In 1941, Seddon introduced a classification of nerve injuries based on three main types of nerve fiber injury and whether there is continuity of the nerve. Usually, however, peripheral nerve injuries are classified in five stages, based on the extent of damage to both the nerve and the surrounding connective tissue, since supporting glial cells may be involved.
Neuroregeneration involves the regrowth or repair of nervous tissues, cells or cell products. Neuroregenerative mechanisms may include generation of new neurons, glia, axons, myelin, or synapses. Neuroregeneration differs between the peripheral nervous system (PNS) and the central nervous system (CNS) by the functional mechanisms involved, especially in the extent and speed of repair. When an axon is damaged, the distal segment undergoes Wallerian degeneration, losing its myelin sheath. The proximal segment can either die by apoptosis or undergo the chromatolytic reaction, which is an attempt at repair. In the CNS, synaptic stripping occurs as glial foot processes invade the dead synapse.
Myelinogenesis is the formation and development of myelin sheaths in the nervous system, typically initiated in late prenatal neurodevelopment and continuing throughout postnatal development. Myelinogenesis continues throughout the lifespan to support learning and memory via neural circuit plasticity as well as remyelination following injury. Successful myelination of axons increases action potential speed by enabling saltatory conduction, which is essential for timely signal conduction between spatially separate brain regions, as well as provides metabolic support to neurons.
A glial scar formation (gliosis) is a reactive cellular process involving astrogliosis that occurs after injury to the central nervous system. As with scarring in other organs and tissues, the glial scar is the body's mechanism to protect and begin the healing process in the nervous system.

Nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) is an enzyme that in humans is encoded by the nmnat1 gene. It is a member of the nicotinamide-nucleotide adenylyltransferases (NMNATs) which catalyze nicotinamide adenine dinucleotide (NAD) synthesis.

Nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) is an enzyme that in humans is encoded by the NMNAT2 gene.
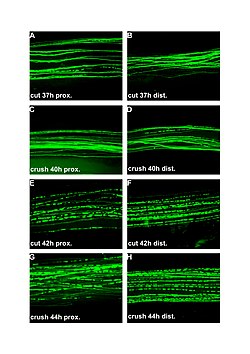
Preferential motor reinnervation (PMR) refers to the tendency of a regenerating axon in the peripheral nervous system (PNS) to reinnervate a motor pathway as opposed to a somatosensory pathway. PMR affects how nerves regenerate and reinnervate within the PNS after surgical procedures or traumatic injuries. It is important to understand in order to further develop axonal regrowth surgical techniques. Further research of preferential motor reinnervation will lead to a better understanding of peripheral nervous system function in the human body regarding cell roles and abilities.

Sterile alpha and TIR motif containing 1 Is an enzyme that in humans is encoded by the SARM1 gene. It is the most evolutionarily conserved member of the Toll/Interleukin receptor-1 (TIR) family. SARM1's TIR domain has intrinsic NADase enzymatic activity that is highly conserved from archaea, plants, nematode worms, fruit flies, and humans. In mammals, SARM1 is highly expressed in neurons, where it resides in both cell bodies and axons, and can be associated with mitochondria.