
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Caroli disease is a rare inherited disorder characterized by cystic dilatation of the bile ducts within the liver. There are two patterns of Caroli disease: focal or simple Caroli disease consists of abnormally widened bile ducts affecting an isolated portion of liver. The second form is more diffuse, and when associated with portal hypertension and congenital hepatic fibrosis, is often referred to as "Caroli syndrome". The underlying differences between the two types are not well understood. Caroli disease is also associated with liver failure and polycystic kidney disease. The disease affects about one in 1,000,000 people, with more reported cases of Caroli syndrome than of Caroli disease.
Multisystem developmental disorder (MSDD) is a term used by Stanley Greenspan to describe children under age 3 who exhibit signs of impaired communication as in autism, but with strong emotional attachments atypical of autism. It is described in the DC:0-3R manual as an optional diagnosis for children under two years of age.

Biliary atresia, also known as extrahepatic ductopenia and progressive obliterative cholangiopathy, is a childhood disease of the liver in which one or more bile ducts are abnormally narrow, blocked, or absent. It can be congenital or acquired. It has an incidence of one in 10,000–15,000 live births in the United States, and a prevalence of one in 16,700 in the British Isles. Biliary atresia is most common in East Asia, with a frequency of one in 5,000.

Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is an autoimmune disease of the liver. It results from a slow, progressive destruction of the small bile ducts of the liver, causing bile and other toxins to build up in the liver, a condition called cholestasis. Further slow damage to the liver tissue can lead to scarring, fibrosis, and eventually cirrhosis.

Alport syndrome is a genetic disorder affecting around 1 in 5,000-10,000 children, characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.

ICF syndrome is a very rare autosomal recessive immune disorder.

Zellweger syndrome is a rare congenital disorder characterized by the reduction or absence of functional peroxisomes in the cells of an individual. It is one of a family of disorders called Zellweger spectrum disorders which are leukodystrophies. Zellweger syndrome is named after Hans Zellweger (1909–1990), a Swiss-American pediatrician, a professor of pediatrics and genetics at the University of Iowa who researched this disorder.
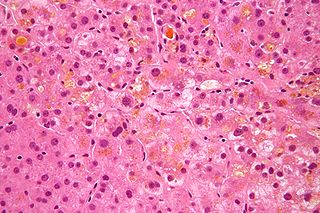
Cholestasis is a condition where the flow of bile from the liver to the duodenum is impaired. The two basic distinctions are:

Dyskeratosis congenita (DKC), also known as Zinsser-Engman-Cole syndrome, is a rare progressive congenital disorder with a highly variable phenotype. The entity was classically defined by the triad of abnormal skin pigmentation, nail dystrophy, and leukoplakia of the oral mucosa, and MDS/AML, but these components do not always occur. DKC is characterized by short telomeres. Some of the manifestations resemble premature ageing and cognitive impairment can be a feature. The disease initially mainly affects the skin, but a major consequence is progressive bone marrow failure which occurs in over 80%, causing early mortality.

Crigler–Najjar syndrome is a rare inherited disorder affecting the metabolism of bilirubin, a chemical formed from the breakdown of the heme in red blood cells. The disorder results in a form of nonhemolytic jaundice, which results in high levels of unconjugated bilirubin and often leads to brain damage in infants. The disorder is inherited in an autosomal recessive manner. The annual incidence is estimated at 1 in 1,000,000.
Nijmegen breakage syndrome (NBS) is a rare autosomal recessive congenital disorder causing chromosomal instability, probably as a result of a defect in the double Holliday junction DNA repair mechanism and/or the synthesis dependent strand annealing mechanism for repairing double strand breaks in DNA.

Apparent mineralocorticoid excess is an autosomal recessive disorder causing hypertension, hypernatremia and hypokalemia. It results from mutations in the HSD11B2 gene, which encodes the kidney isozyme of 11β-hydroxysteroid dehydrogenase type 2. In an unaffected individual, this isozyme inactivates circulating cortisol to the less active metabolite cortisone. The inactivating mutation leads to elevated local concentrations of cortisol in the aldosterone sensitive tissues like the kidney. Cortisol at high concentrations can cross-react and activate the mineralocorticoid receptor due to the non-selectivity of the receptor, leading to aldosterone-like effects in the kidney. This is what causes the hypokalemia, hypertension, and hypernatremia associated with the syndrome. Patients often present with severe hypertension and end-organ changes associated with it like left ventricular hypertrophy, retinal, renal and neurological vascular changes along with growth retardation and failure to thrive. In serum both aldosterone and renin levels are low.
Progressive familial intrahepatic cholestasis (PFIC) is a group of familial cholestatic conditions caused by defects in biliary epithelial transporters. The clinical presentation usually occurs first in childhood with progressive cholestasis. This usually leads to failure to thrive, cirrhosis, and the need for liver transplantation.

Hajdu–Cheney syndrome, also called acroosteolysis with osteoporosis and changes in skull and mandible, arthrodentoosteodysplasia and Cheney syndrome, is an extremely rare autosomal dominant congenital disorder of the connective tissue characterized by severe and excessive bone resorption leading to osteoporosis and a wide range of other possible symptoms. Mutations in the NOTCH2 gene, identified in 2011, cause HCS. HCS is so rare that only about 50 cases have been reported worldwide since the discovery of the syndrome in 1948
Neonatal cholestasis refers to elevated levels of conjugated bilirubin identified in newborn infants within the first few months of life. Conjugated hyperbilirubinemia is clinically defined as >20% of total serum bilirubin or conjugated bilirubin concentration greater than 1.0 mg/dL regardless of total serum bilirubin concentration. The differential diagnosis for neonatal cholestasis can vary extensively. However, the underlying disease pathology is caused by improper transport and/or defects in excretion of bile from hepatocytes leading to an accumulation of conjugated bilirubin in the body. Generally, symptoms associated with neonatal cholestasis can vary based on the underlying cause of the disease. However, most infants affected will present with jaundice, scleral icterus, failure to thrive, acholic or pale stools, and dark urine.

Jagged1 (JAG1) is one of five cell surface proteins (ligands) that interact with four receptors in the mammalian Notch signaling pathway. The Notch signaling pathway is a highly conserved pathway that functions to establish and regulate cell fate decisions in many organ systems. Once the JAG1-NOTCH (receptor-ligand) interactions take place, a cascade of proteolytic cleavages is triggered resulting in activation of the transcription for downstream target genes. Located on human chromosome 20, the JAG1 gene is expressed in multiple organ systems in the body and causes the autosomal dominant disorder Alagille syndrome (ALGS) resulting from loss of function mutations within the gene. JAG1 has also been designated as CD339.

Renal–hepatic–pancreatic dysplasia is an autosomal recessive congenital disorder characterized by pancreatic fibrosis, renal dysplasia and hepatic dysgenesis. An association with NPHP3 has been described. It was characterized in 1959.
Vanishing bile duct syndrome is a loose collection of diseases which leads to the injury to hepatic bile ducts and eventual ductopenia.
Mitochondrial DNA depletion syndrome, or Alper's disease, is any of a group of autosomal recessive disorders that cause a significant drop in mitochondrial DNA in affected tissues. Symptoms can be any combination of myopathic, hepatopathic, or encephalomyopathic. These syndromes affect tissue in the muscle, liver, or both the muscle and brain, respectively. The condition is typically fatal in infancy and early childhood, though some have survived to their teenage years with the myopathic variant and some have survived into adulthood with the SUCLA2 encephalomyopathic variant. There is currently no curative treatment for any form of MDDS, though some preliminary treatments have shown a reduction in symptoms.

