
Schizophrenia is a mental disorder characterized by reoccurring episodes of psychosis that are correlated with a general misperception of reality. Other common signs include hallucinations, delusions, disorganized thinking, social withdrawal, and flat affect. Symptoms develop gradually and typically begin during young adulthood and are never resolved. There is no objective diagnostic test; diagnosis is based on observed behavior, a psychiatric history that includes the person's reported experiences, and reports of others familiar with the person. For a diagnosis of schizophrenia, the described symptoms need to have been present for at least six months or one month. Many people with schizophrenia have other mental disorders, especially substance use disorders, depressive disorders, anxiety disorders, and obsessive–compulsive disorder.

Reelin, encoded by the RELN gene, is a large secreted extracellular matrix glycoprotein that helps regulate processes of neuronal migration and positioning in the developing brain by controlling cell–cell interactions. Besides this important role in early development, reelin continues to work in the adult brain. It modulates synaptic plasticity by enhancing the induction and maintenance of long-term potentiation. It also stimulates dendrite and dendritic spine development and regulates the continuing migration of neuroblasts generated in adult neurogenesis sites like the subventricular and subgranular zones. It is found not only in the brain but also in the liver, thyroid gland, adrenal gland, fallopian tube, breast and in comparatively lower levels across a range of anatomical regions.
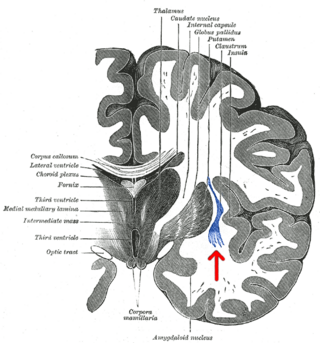
The claustrum is a thin sheet of neurons and supporting glial cells, that connects to the cerebral cortex and subcortical regions including the amygdala, hippocampus and thalamus of the brain. It is located between the insular cortex laterally and the putamen medially, encased by the extreme and external capsules respectively. Blood to the claustrum is supplied by the middle cerebral artery. It is considered to be the most densely connected structure in the brain, and thus hypothesized to allow for the integration of various cortical inputs such as vision, sound and touch, into one experience. Other hypotheses suggest that the claustrum plays a role in salience processing, to direct attention towards the most behaviorally relevant stimuli amongst the background noise. The claustrum is difficult to study given the limited number of individuals with claustral lesions and the poor resolution of neuroimaging.
The dopamine hypothesis of schizophrenia or the dopamine hypothesis of psychosis is a model that attributes the positive symptoms of schizophrenia to a disturbed and hyperactive dopaminergic signal transduction. The model draws evidence from the observation that a large number of antipsychotics have dopamine-receptor antagonistic effects. The theory, however, does not posit dopamine overabundance as a complete explanation for schizophrenia. Rather, the overactivation of D2 receptors, specifically, is one effect of the global chemical synaptic dysregulation observed in this disorder.

Dopaminergic pathways in the human brain are involved in both physiological and behavioral processes including movement, cognition, executive functions, reward, motivation, and neuroendocrine control. Each pathway is a set of projection neurons, consisting of individual dopaminergic neurons.

Dizocilpine (INN), also known as MK-801, is a pore blocker of the NMDA receptor, a glutamate receptor, discovered by a team at Merck in 1982. Glutamate is the brain's primary excitatory neurotransmitter. The channel is normally blocked with a magnesium ion and requires depolarization of the neuron to remove the magnesium and allow the glutamate to open the channel, causing an influx of calcium, which then leads to subsequent depolarization. Dizocilpine binds inside the ion channel of the receptor at several of PCP's binding sites thus preventing the flow of ions, including calcium (Ca2+), through the channel. Dizocilpine blocks NMDA receptors in a use- and voltage-dependent manner, since the channel must open for the drug to bind inside it. The drug acts as a potent anti-convulsant and probably has dissociative anesthetic properties, but it is not used clinically for this purpose because of the discovery of brain lesions, called Olney's lesions (see below), in laboratory rats. Dizocilpine is also associated with a number of negative side effects, including cognitive disruption and psychotic-spectrum reactions. It inhibits the induction of long term potentiation and has been found to impair the acquisition of difficult, but not easy, learning tasks in rats and primates. Because of these effects of dizocilpine, the NMDA receptor pore blocker ketamine is used instead as a dissociative anesthetic in human medical procedures. While ketamine may also trigger temporary psychosis in certain individuals, its short half-life and lower potency make it a much safer clinical option. However, dizocilpine is the most frequently used uncompetitive NMDA receptor antagonist in animal models to mimic psychosis for experimental purposes.

The metabotropic glutamate receptors, or mGluRs, are a type of glutamate receptor that are active through an indirect metabotropic process. They are members of the group C family of G-protein-coupled receptors, or GPCRs. Like all glutamate receptors, mGluRs bind with glutamate, an amino acid that functions as an excitatory neurotransmitter.

Glutamate receptors are synaptic and non synaptic receptors located primarily on the membranes of neuronal and glial cells. Glutamate is abundant in the human body, but particularly in the nervous system and especially prominent in the human brain where it is the body's most prominent neurotransmitter, the brain's main excitatory neurotransmitter, and also the precursor for GABA, the brain's main inhibitory neurotransmitter. Glutamate receptors are responsible for the glutamate-mediated postsynaptic excitation of neural cells, and are important for neural communication, memory formation, learning, and regulation.
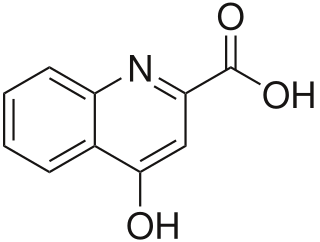
Kynurenic acid is a product of the normal metabolism of amino acid L-tryptophan. It has been shown that kynurenic acid possesses neuroactive activity. It acts as an antiexcitotoxic and anticonvulsant, most likely through acting as an antagonist at excitatory amino acid receptors. Because of this activity, it may influence important neurophysiological and neuropathological processes. As a result, kynurenic acid has been considered for use in therapy in certain neurobiological disorders. Conversely, increased levels of kynurenic acid have also been linked to certain pathological conditions.

Chandelier cells or chandelier neurons are a subset of GABAergic cortical interneurons. They are described as parvalbumin-containing and fast-spiking to distinguish them from other subtypes of GABAergic neurons, although some studies have suggested that only a subset of chandelier cells test positive for parvalbumin by immunostaining. The name comes from the specific shape of their axon arbors, with the terminals forming distinct arrays called "cartridges". The cartridges are immunoreactive to an isoform of the GABA membrane transporter, GAT-1, and this serves as their identifying feature. GAT-1 is involved in the process of GABA reuptake into nerve terminals, thus helping to terminate its synaptic activity. Chandelier neurons synapse exclusively to the axonal initial segment of pyramidal neurons, near the site where action potential is generated. It is believed that they provide inhibitory input to the pyramidal neurons, but there is data showing that in some circumstances the GABA from chandelier neurons could be excitatory.
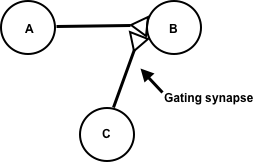
Synaptic gating is the ability of neural circuits to gate inputs by either suppressing or facilitating specific synaptic activity. Selective inhibition of certain synapses has been studied thoroughly, and recent studies have supported the existence of permissively gated synaptic transmission. In general, synaptic gating involves a mechanism of central control over neuronal output. It includes a sort of gatekeeper neuron, which has the ability to influence transmission of information to selected targets independently of the parts of the synapse upon which it exerts its action.

Protein phosphatase 1 regulatory subunit 1B (PPP1R1B), also known as dopamine- and cAMP-regulated neuronal phosphoprotein (DARPP-32), is a protein that in humans is encoded by the PPP1R1B gene.
The glutamate hypothesis of schizophrenia models the subset of pathologic mechanisms of schizophrenia linked to glutamatergic signaling. The hypothesis was initially based on a set of clinical, neuropathological, and, later, genetic findings pointing at a hypofunction of glutamatergic signaling via NMDA receptors. While thought to be more proximal to the root causes of schizophrenia, it does not negate the dopamine hypothesis, and the two may be ultimately brought together by circuit-based models. The development of the hypothesis allowed for the integration of the GABAergic and oscillatory abnormalities into the converging disease model and made it possible to discover the causes of some disruptions.
Scientific studies have found that different brain areas show altered activity in humans with major depressive disorder (MDD), and this has encouraged advocates of various theories that seek to identify a biochemical origin of the disease, as opposed to theories that emphasize psychological or situational causes. Factors spanning these causative groups include nutritional deficiencies in magnesium, vitamin D, and tryptophan with situational origin but biological impact. Several theories concerning the biologically based cause of depression have been suggested over the years, including theories revolving around monoamine neurotransmitters, neuroplasticity, neurogenesis, inflammation and the circadian rhythm. Physical illnesses, including hypothyroidism and mitochondrial disease, can also trigger depressive symptoms.
The biology of obsessive–compulsive disorder (OCD) refers biologically based theories about the mechanism of OCD. Cognitive models generally fall into the category of executive dysfunction or modulatory control. Neuroanatomically, functional and structural neuroimaging studies implicate the prefrontal cortex (PFC), basal ganglia (BG), insula, and posterior cingulate cortex (PCC). Genetic and neurochemical studies implicate glutamate and monoamine neurotransmitters, especially serotonin and dopamine.

Visual processing abnormalities in schizophrenia are commonly found, and contribute to poor social function.

Hypofrontality is a state of decreased cerebral blood flow (CBF) in the prefrontal cortex of the brain. Hypofrontality is symptomatic of several neurological medical conditions, such as schizophrenia, attention deficit hyperactivity disorder (ADHD), bipolar disorder, and major depressive disorder. This condition was initially described by Ingvar and Franzén in 1974, through the use of xenon blood flow technique with 32 detectors to image the brains of patients with schizophrenia. This finding was confirmed in subsequent studies using the improved spatial resolution of positron emission tomography with the fluorodeoxyglucose (18F-FDG) tracer. Subsequent neuroimaging work has shown that the decreases in prefrontal CBF are localized to the medial, lateral, and orbital portions of the prefrontal cortex. Hypofrontality is thought to contribute to the negative symptoms of schizophrenia.

Bipolar disorder is an affective disorder characterized by periods of elevated and depressed mood. The cause and mechanism of bipolar disorder is not yet known, and the study of its biological origins is ongoing. Although no single gene causes the disorder, a number of genes are linked to increase risk of the disorder, and various gene environment interactions may play a role in predisposing individuals to developing bipolar disorder. Neuroimaging and postmortem studies have found abnormalities in a variety of brain regions, and most commonly implicated regions include the ventral prefrontal cortex and amygdala. Dysfunction in emotional circuits located in these regions have been hypothesized as a mechanism for bipolar disorder. A number of lines of evidence suggests abnormalities in neurotransmission, intracellular signalling, and cellular functioning as possibly playing a role in bipolar disorder.

Margarita Behrens is a neuroscientist and biochemist. She is currently an associate professor at the Salk Institute for Biological Studies where her lab studies the impact of oxidative stress on the post-natal brain through probing the biology of fast-spiking parvalbumin interneurons in models of schizophrenia.

Bita Moghaddam is an Iranian-American neuroscientist and author. She is currently the Ruth Matarazzo Professor of Behavioral Neuroscience at Oregon Health & Science University. Moghaddam investigates the neuronal processes underlying emotion and cognition as a first step to designing strategies to treat and prevent brain illnesses.
