
A thrombus, colloquially called a blood clot, is the final product of the blood coagulation step in hemostasis. There are two components to a thrombus: aggregated platelets and red blood cells that form a plug, and a mesh of cross-linked fibrin protein. The substance making up a thrombus is sometimes called cruor. A thrombus is a healthy response to injury intended to stop and prevent further bleeding, but can be harmful in thrombosis, when a clot obstructs blood flow through healthy blood vessels in the circulatory system.

Platelets or thrombocytes are a component of blood whose function is to react to bleeding from blood vessel injury by clumping, thereby initiating a blood clot. Platelets have no cell nucleus; they are fragments of cytoplasm derived from the megakaryocytes of the bone marrow or lung, which then enter the circulation. Platelets are found only in mammals, whereas in other vertebrates, thrombocytes circulate as intact mononuclear cells.

Coagulation, also known as clotting, is the process by which blood changes from a liquid to a gel, forming a blood clot. It potentially results in hemostasis, the cessation of blood loss from a damaged vessel, followed by repair. The mechanism of coagulation involves activation, adhesion and aggregation of platelets, as well as deposition and maturation of fibrin.

Disseminated intravascular coagulation (DIC) is a condition in which blood clots form throughout the body, blocking small blood vessels. Symptoms may include chest pain, shortness of breath, leg pain, problems speaking, or problems moving parts of the body. As clotting factors and platelets are used up, bleeding may occur. This may include blood in the urine, blood in the stool, or bleeding into the skin. Complications may include organ failure.

Fibrinogen is a glycoprotein complex, produced in the liver, that circulates in the blood of all vertebrates. During tissue and vascular injury, it is converted enzymatically by thrombin to fibrin and then to a fibrin-based blood clot. Fibrin clots function primarily to occlude blood vessels to stop bleeding. Fibrin also binds and reduces the activity of thrombin. This activity, sometimes referred to as antithrombin I, limits clotting. Fibrin also mediates blood platelet and endothelial cell spreading, tissue fibroblast proliferation, capillary tube formation, and angiogenesis and thereby promotes revascularization and wound healing.

Thrombin is a serine protease, an enzyme that, in humans, is encoded by the F2 gene.
D-dimer is a dimer that is a fibrin degradation product, a small protein fragment present in the blood after a blood clot is degraded by fibrinolysis. It is so named because it contains two D fragments of the fibrin protein joined by a cross-link, hence forming a protein dimer.
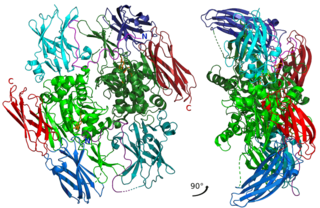
Factor XIII or fibrin stabilizing factor is a zymogen found in blood of humans and some other animals. It is activated by thrombin to factor XIIIa. Factor XIIIa is an enzyme of the blood coagulation system that crosslinks fibrin. Deficiency of XIII worsens clot stability and increases bleeding tendency.

Hirudin is a naturally occurring peptide in the salivary glands of blood-sucking leeches that has a blood anticoagulant property. This is essential for the leeches' habit of feeding on blood, since it keeps a host's blood flowing after the worm's initial puncture of the skin.
In biochemistry and medicine, glycoprotein IIb/IIIa is an integrin complex found on platelets. It is a transmembrane receptor for fibrinogen and von Willebrand factor, and aids platelet activation. The complex is formed via calcium-dependent association of gpIIb and gpIIIa, a required step in normal platelet aggregation and endothelial adherence. Platelet activation by ADP leads to the aforementioned conformational change in platelet gpIIb/IIIa receptors that induces binding to fibrinogen. The gpIIb/IIIa receptor is a target of several drugs including abciximab, eptifibatide, and tirofiban.
Cytochalasins are fungal metabolites that have the ability to bind to actin filaments and block polymerization and the elongation of actin. As a result of the inhibition of actin polymerization, cytochalasins can change cellular morphology, inhibit cellular processes such as cell division, and even cause cells to undergo apoptosis. Cytochalasins have the ability to permeate cell membranes, prevent cellular translocation and cause cells to enucleate. Cytochalasins can also have an effect on other aspects of biological processes unrelated to actin polymerization. For example, cytochalasin A and cytochalasin B can also inhibit the transport of monosaccharides across the cell membrane, cytochalasin H has been found to regulate plant growth, cytochalasin D inhibits protein synthesis and cytochalasin E prevents angiogenesis.

Batroxobin, also known as reptilase, is a snake venom enzyme with Venombin A activity produced by Bothrops atrox and Bothrops moojeni, venomous species of pit viper found east of the Andes in South America. It is a hemotoxin which acts as a serine protease similarly to thrombin, and has been the subject of many medical studies as a replacement of thrombin. Different enzymes, isolated from different species of Bothrops, have been called batroxobin, but unless stated otherwise, this article covers the batroxobin produced by B. moojeni, as this is the most studied variety.
The dysfibrinogenemias consist of three types of fibrinogen disorders in which a critical blood clotting factor, fibrinogen, circulates at normal levels but is dysfunctional. Congenital dysfibrinogenemia is an inherited disorder in which one of the parental genes produces an abnormal fibrinogen. This fibrinogen interferes with normal blood clotting and/or lysis of blood clots. The condition therefore may cause pathological bleeding and/or thrombosis. Acquired dysfibrinogenemia is a non-hereditary disorder in which fibrinogen is dysfunctional due to the presence of liver disease, autoimmune disease, a plasma cell dyscrasias, or certain cancers. It is associated primarily with pathological bleeding. Hereditary fibrinogen Aα-Chain amyloidosis is a sub-category of congenital dysfibrinogenemia in which the dysfunctional fibrinogen does not cause bleeding or thrombosis but rather gradually accumulates in, and disrupts the function of, the kidney.

Fibrinogen gamma chain, also known as fibrinogen gamma gene (FGG), is a human gene found on chromosome 3.
Platelet membrane glycoproteins are surface glycoproteins found on platelets (thrombocytes) which play a key role in hemostasis. When the blood vessel wall is damaged, platelet membrane glycoproteins interact with the extracellular matrix.

Fibrinogen alpha chain is a protein that in humans is encoded by the FGA gene.
Blood clotting tests are the tests used for diagnostics of the hemostasis system. Coagulometer is the medical laboratory analyzer used for testing of the hemostasis system. Modern coagulometers realize different methods of activation and observation of development of blood clots in blood or in blood plasma.
The haemostatic system involves the interaction of proteins in the blood, the blood vessel wall and the flow of blood to control bleeding and blood clotting. Developmental Haemostasis is a term that represents the maturation of the haemostatic system from birth to adulthood. There are differences in the concentration, structure and activity of many proteins involved in blood clotting. These changes play an important role in physiological development and are important in providing appropriate diagnosis and treatment of bleeding and clotting disorders. The age-specific differences in the blood clotting system may contribute to the fact that children are less prone to developing thrombosis compared to adults.
The platelet plug, also known as the hemostatic plug or platelet thrombus, is an aggregation of platelets formed during early stages of hemostasis in response to one or more injuries to blood vessel walls. After platelets are recruited and begin to accumulate around the breakage, their “sticky” nature allows them to adhere to each other. This forms a platelet plug, which prevents more blood from leaving the body as well as any outside contaminants from getting in. The plug provides a temporary blockage of the break in the vasculature. As such, platelet plug formation occurs after vasoconstriction of the blood vessels but before the creation of the fibrin mesh clot, which is the more permanent solution to the injury. The result of the platelet plug formation is the coagulation of blood. It can also be referred to as primary hemostasis.
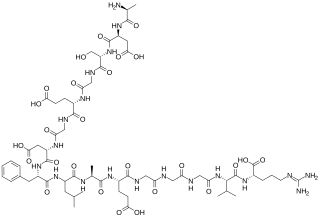
The fibrinopeptides, fibrinopeptide A (FpA) and fibrinopeptide B (FpB), are peptides which are located in the central region of the fibrous glycoprotein fibrinogen and are cleaved by the enzyme thrombin to convert fibrinogen into covalently-linked fibrin monomers. The N-terminal FpA is cleaved from the Aα chains of fibrinogen and FpB from the Bβ chains of fibrinogen, with FpA released before FpB. Subsequent to their formation, fibrin monomers are converted to cross-linked fibrin polymers by the action of thrombin-activated factor XIII, and these fibrin polymers form the backbone of a thrombus. Hence, the fibrinopeptides are sensitive markers of fibrinogenesis, thrombin activity, and coagulation.




