
Turner syndrome (TS), also known as 45,X, or 45,X0, is a genetic condition in which a female is partially or completely missing an X chromosome. Signs and symptoms vary among those affected. Often, a short and webbed neck, low-set ears, low hairline at the back of the neck, short stature, and swollen hands and feet are seen at birth. Typically, those affected do not develop menstrual periods and breasts without hormone treatment and are unable to have children without reproductive technology. Heart defects, diabetes, and low thyroid hormone occur more frequently than average in the disorder. Most people with TS have normal intelligence; however, many have problems with spatial visualization that may be needed for mathematics. Vision and hearing problems also occur more often than average.

Ovarian cancer is a cancer that forms in or on an ovary. It results in abnormal cells that have the ability to invade or spread to other parts of the body. When this process begins, there may be no or only vague symptoms. Symptoms become more noticeable as the cancer progresses. These symptoms may include bloating, pelvic pain, abdominal swelling, constipation, and loss of appetite, among others. Common areas to which the cancer may spread include the lining of the abdomen, lymph nodes, lungs, and liver.

XY gonadal dysgenesis, also known as Swyer syndrome, is a type of hypogonadism in a person whose karyotype is 46,XY. Though they typically have normal female external genitalia, the person has functionless gonads, fibrous tissue termed "streak gonads", and if left untreated, will not experience puberty. Such gonads are typically surgically removed. The typical medical treatment is hormone replacement therapy. The syndrome was named after Gerald Swyer, an endocrinologist based in London.
Delayed puberty is when a person lacks or has incomplete development of specific sexual characteristics past the usual age of onset of puberty. The person may have no physical or hormonal signs that puberty has begun. In the United States, girls are considered to have delayed puberty if they lack breast development by age 13 or have not started menstruating by age 16. Boys are considered to have delayed puberty if they lack enlargement of the testicles by age 14. Delayed puberty affects about 2% of adolescents.

A dysgerminoma is a type of germ cell tumor; it usually is malignant and usually occurs in the ovary.

Germ cell tumor (GCT) is a neoplasm derived from germ cells. Germ-cell tumors can be cancerous or benign. Germ cells normally occur inside the gonads. GCTs that originate outside the gonads may be birth defects resulting from errors during development of the embryo.
A small supernumerary marker chromosome (sSMC) is an abnormal extra chromosome. It contains copies of parts of one or more normal chromosomes and like normal chromosomes is located in the cell's nucleus, is replicated and distributed into each daughter cell during cell division, and typically has genes which may be expressed. However, it may also be active in causing birth defects and neoplasms. The sSMC's small size makes it virtually undetectable using classical cytogenetic methods: the far larger DNA and gene content of the cell's normal chromosomes obscures those of the sSMC. Newer molecular techniques such as fluorescence in situ hybridization, next generation sequencing, comparative genomic hybridization, and highly specialized cytogenetic G banding analyses are required to study it. Using these methods, the DNA sequences and genes in sSMCs are identified and help define as well as explain any effect(s) it may have on individuals.
XX gonadal dysgenesis is a type of female hypogonadism in which no functional ovaries are present to induce puberty in an otherwise normal girl whose karyotype is found to be 46,XX. With nonfunctional streak ovaries, she is low in estrogen levels (hypoestrogenic) and has high levels of FSH and LH. Estrogen and progesterone therapy is usually then commenced.
Gonadal dysgenesis is classified as any congenital developmental disorder of the reproductive system in the male or female. It is the atypical development of the gonads in an embryo, with reproductive tissue replaced with functionless, fibrous tissue, termed streak gonads. Streak gonads are a form of aplasia, resulting in hormonal failure that manifests as sexual infantism and infertility, with no initiation of puberty and secondary sex characteristics.
A germinoma is a type of germ-cell tumor, which is not differentiated upon examination. It may be benign or malignant.

Endocrine diseases are disorders of the endocrine system. The branch of medicine associated with endocrine disorders is known as endocrinology.

Disorders of sex development (DSDs), also known as differences in sex development, diverse sex development and variations in sex characteristics (VSC), are congenital conditions affecting the reproductive system, in which development of chromosomal, gonadal, or anatomical sex is atypical.

The steroidogenic factor 1 (SF-1) protein is a transcription factor involved in sex determination by controlling activity of genes related to the reproductive glands or gonads and adrenal glands. This protein is encoded by the NR5A1 gene, a member of the nuclear receptor subfamily, located on the long arm of chromosome 9 at position 33.3. It was originally identified as a regulator of genes encoding cytochrome P450 steroid hydroxylases, however, further roles in endocrine function have since been discovered.
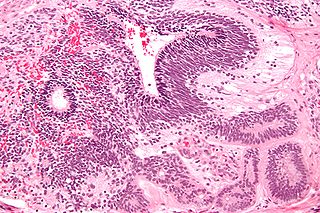
An immature teratoma is a teratoma that contains anaplastic immature elements, and is often synonymous with malignant teratoma. A teratoma is a tumor of germ cell origin, containing tissues from more than one germ cell line, It can be ovarian or testicular in its origin. and are almost always benign. An immature teratoma is thus a very rare tumor, representing 1% of all teratomas, 1% of all ovarian cancers, and 35.6% of malignant ovarian germ cell tumors. It displays a specific age of incidence, occurring most frequently in the first two decades of life and almost never after menopause. Unlike a mature cystic teratoma, an immature teratoma contains immature or embryonic structures. It can coexist with mature cystic teratomas and can constitute of a combination of both adult and embryonic tissue. The most common symptoms noted are abdominal distension and masses. Prognosis and treatment options vary and largely depend on grade, stage and karyotype of the tumor itself.
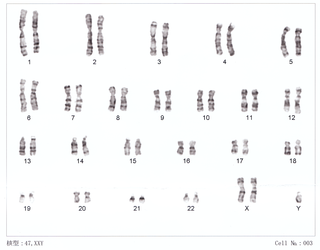
Klinefelter syndrome (KS), also known as 47,XXY, is a syndrome where a male has an additional copy of the X chromosome. The primary features are infertility and small, poorly functioning testicles. Often, symptoms are subtle and subjects do not realize they are affected. Sometimes, symptoms are more evident and may include weaker muscles, greater height, poor motor coordination, less body hair, breast growth, and less interest in sex. Often, these symptoms are noticed only at puberty. Intelligence is usually normal, but reading difficulties and problems with speech are more common.

Complete androgen insensitivity syndrome (CAIS) is an AIS condition that results in the complete inability of the cell to respond to androgens. As such, the insensitivity to androgens is only clinically significant when it occurs in individuals with a Y chromosome or, more specifically, an SRY gene. The unresponsiveness of the cell to the presence of androgenic hormones prevents the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty, but does allow, without significant impairment, female genital and sexual development in those with the condition.
46,XX/46,XY is an exceptionally rare chimeric genetic condition characterized by the presence of some cells that express a 46,XX karyotype and some cells that express a 46,XY karyotype in a single human being. The cause of the condition lies in utero with the aggregation of two distinct blastocysts or zygotes into a single embryo, which subsequently leads to the development of a single individual with two distinct cell lines, instead of a pair of fraternal twins. 46,XX/46,XY chimeras are the result of the merging of two non-identical twins. This is not to be confused with mosaicism or hybridism, neither of which are chimeric conditions. Since individuals with the condition have two cell lines of the opposite sex, it can also be considered an intersex condition.
45,X/46,XY mosaicism, also known as X0/XY mosaicism and mixed gonadal dysgenesis, is a rare mutation of sex development in humans associated with sex chromosome aneuploidy and mosaicism of the Y chromosome. This is called a mosaic karyotype because, like tiles in mosaic floors or walls, there is more than one type of cell. It is a fairly rare chromosomal disorder, with an estimated incidence rate of about 1 in 15,000 live births.
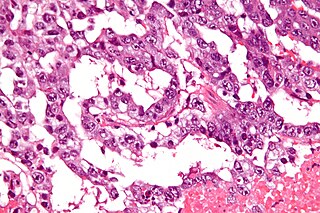
An Extracranial Germ-Cell Tumor (EGCT) occurs in the abnormal growth of germ cells in the gonads and the areas other than the brain via tissue, lymphatic system, or circulatory system. The tumor can be benign or malignant (cancerous) by its growth rate. According to the National Cancer Institute and St. Jude Children's Research Hospital, the chance of children who are under 15 years old having EGCTs is 3%, in comparison to adolescents, a possibility of 14% with aged 15 to 19 can have EGCTs. There is no obvious cut point in between children and adolescents. However, common cut points in researches are 11 years old and 15 years old.
Ovarian germ cell tumors (OGCTs) are heterogeneous tumors that are derived from the primitive germ cells of the embryonic gonad, which accounts for about 2.6% of all ovarian malignancies. There are four main types of OGCTs, namely dysgerminomas, yolk sac tumor, teratoma, and choriocarcinoma.
