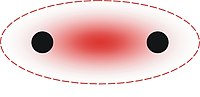
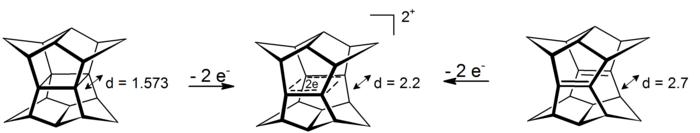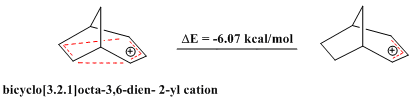In theoretical chemistry, a conjugated system is a system of connected p-orbitals with delocalized electrons in a molecule, which in general lowers the overall energy of the molecule and increases stability. It is conventionally represented as having alternating single and multiple bonds. Lone pairs, radicals or carbenium ions may be part of the system, which may be cyclic, acyclic, linear or mixed. The term "conjugated" was coined in 1899 by the German chemist Johannes Thiele.

In chemistry, aromaticity means a molecule has a cyclic (ring-shaped) structure with pi bonds in resonance. Aromatic rings give increased stability compared to saturated compounds having single bonds, and other geometric or connective non-cyclic arrangements with the same set of atoms. Aromatic rings are very stable and do not break apart easily. Organic compounds that are not aromatic are classified as aliphatic compounds—they might be cyclic, but only aromatic rings have enhanced stability. The term aromaticity with this meaning is historically related to the concept of having an aroma, but is a distinct property from that meaning.
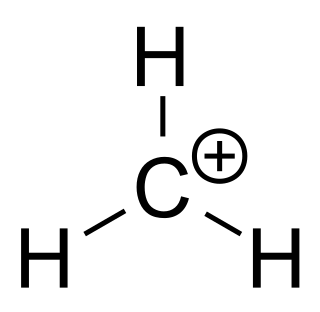
A carbocation is an ion with a positively charged carbon atom. Among the simplest examples are the methenium CH+
3, methanium CH+
5 and vinyl C
2H+
3 cations. Occasionally, carbocations that bear more than one positively charged carbon atom are also encountered.
In chemistry, resonance, also called mesomerism, is a way of describing bonding in certain molecules or polyatomic ions by the combination of several contributing structures into a resonance hybrid in valence bond theory. It has particular value for analyzing delocalized electrons where the bonding cannot be expressed by one single Lewis structure. It is considered as the accurate structure for a compound.
3-Methylenecyclopropene, also called methylenecyclopropene or triafulvene, is a hydrocarbon with chemical formula C4H4. It is a colourless gas that polymerizes readily as a liquid or in solution but is stable as a gas. This highly strained and reactive molecule was synthesized and characterized for the first time in 1984, and has been the subject of considerable experimental and theoretical interest. It is an example of a cross-conjugated alkene, being composed of cyclopropene with an exocyclic double bond attached.
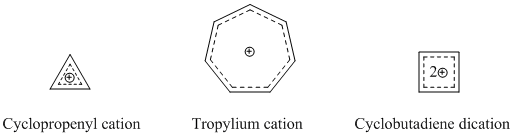
In organic chemistry, Hückel's rule predicts that a planar ring molecule will have aromatic properties if it has 4n + 2 π electrons, where n is a non-negative integer. The quantum mechanical basis for its formulation was first worked out by physical chemist Erich Hückel in 1931. The succinct expression as the 4n + 2 rule has been attributed to W. v. E. Doering (1951), although several authors were using this form at around the same time.
Antiaromaticity is a chemical property of a cyclic molecule with a π electron system that has higher energy, i.e., it is less stable due to the presence of 4n delocalised electrons in it, as opposed to aromaticity. Unlike aromatic compounds, which follow Hückel's rule and are highly stable, antiaromatic compounds are highly unstable and highly reactive. To avoid the instability of antiaromaticity, molecules may change shape, becoming non-planar and therefore breaking some of the π interactions. In contrast to the diamagnetic ring current present in aromatic compounds, antiaromatic compounds have a paramagnetic ring current, which can be observed by NMR spectroscopy.
In molecular geometry, bond length or bond distance is defined as the average distance between nuclei of two bonded atoms in a molecule. It is a transferable property of a bond between atoms of fixed types, relatively independent of the rest of the molecule.
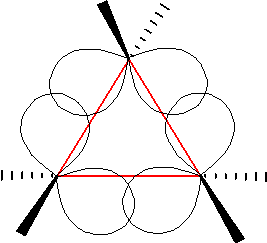
In organic chemistry, a bent bond, also known as a banana bond, is a type of covalent chemical bond with a geometry somewhat reminiscent of a banana. The term itself is a general representation of electron density or configuration resembling a similar "bent" structure within small ring molecules, such as cyclopropane (C3H6) or as a representation of double or triple bonds within a compound that is an alternative to the sigma and pi bond model.

Cyclooctadecanonaene or [18]annulene is an organic compound with chemical formula C
18H
18. It belongs to the class of highly conjugated compounds known as annulenes and is aromatic. The usual isomer that [18]annulene refers to is the most stable one, containing six interior hydrogens and twelve exterior ones, with the nine formal double bonds in the cis,trans,trans,cis,trans,trans,cis,trans,trans configuration. It is reported to be a red-brown crystalline solid.
A carbenium ion is a positive ion with the structure RR′R″C+, that is, a chemical species with carbon atom having three covalent bonds, and it bears a +1 formal charge. But IUPAC confuses coordination number with valence, incorrectly considering carbon in carbenium as trivalent.
An arenium ion in organic chemistry is a cyclohexadienyl cation that appears as a reactive intermediate in electrophilic aromatic substitution. For historic reasons this complex is also called a Wheland intermediate, after American chemist George Willard Wheland (1907–1976). They are also called sigma complexes. The smallest arenium ion is the benzenium ion, which is protonated benzene.
In organic chemistry, the term 2-norbornyl cation describes one of the three carbocations formed from derivatives of norbornane. Though 1-norbornyl and 7-norbornyl cations have been studied, the most extensive studies and vigorous debates have been centered on the exact structure of the 2-norbornyl cation.

An aromatic ring current is an effect observed in aromatic molecules such as benzene and naphthalene. If a magnetic field is directed perpendicular to the plane of the aromatic system, a ring current is induced in the delocalized π electrons of the aromatic ring. This is a direct consequence of Ampère's law; since the electrons involved are free to circulate, rather than being localized in bonds as they would be in most non-aromatic molecules, they respond much more strongly to the magnetic field.
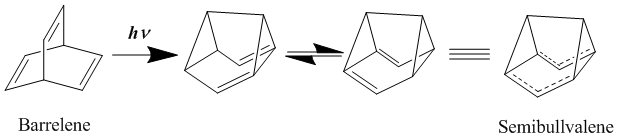
Bullvalene is a hydrocarbon with the chemical formula C10H10. The molecule has a cage-like structure formed by the fusion of one cyclopropane and three cyclohepta-1,4-diene rings. Bullvalene is unusual as an organic molecule due to the C−C and C=C bonds forming and breaking rapidly on the NMR timescale; this property makes it a fluxional molecule.
In chemistry, an oxocarbon anion is a negative ion consisting solely of carbon and oxygen atoms, and therefore having the general formula C
xOn−
y for some integers x, y, and n.
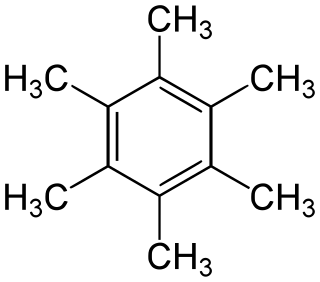
Hexamethylbenzene, also known as mellitene, is a hydrocarbon with the molecular formula C12H18 and the condensed structural formula C6(CH3)6. It is an aromatic compound and a derivative of benzene, where benzene's six hydrogen atoms have each been replaced by a methyl group. In 1929, Kathleen Lonsdale reported the crystal structure of hexamethylbenzene, demonstrating that the central ring is hexagonal and flat and thereby ending an ongoing debate about the physical parameters of the benzene system. This was a historically significant result, both for the field of X-ray crystallography and for understanding aromaticity.

A pyramidal carbocation is a type of carbocation with a specific configuration. This ion exists as a third class, besides the classical and non-classical ions. In these ions, a single carbon atom hovers over a four- or five-sided polygon, in effect forming a pyramid. The four-sided pyramidal ion will carry a charge of 1+, and the five-sided pyramid will carry 2+. In the images, the black spot on the vertical line represents the hovering carbon atom.
Hydrogen-bridged cations are a type of charged species in which a hydrogen atom is simultaneously bonded to two atoms through partial sigma bonds. While best observable in the presence of superacids at room temperature, spectroscopic evidence has suggested that hydrogen-bridged cations exist in ordinary solvents. These ions have been the subject of debate as they constitute a type of charged species of uncertain electronic structure.
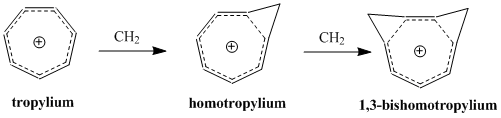
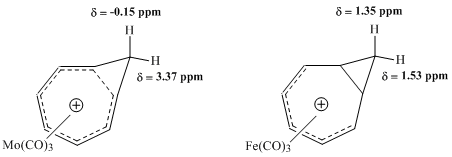
Bicycloaromaticity in chemistry is an extension of the concept of homoaromaticity with two aromatic ring currents situated in a non-planar molecule and sharing the same electrons. The concept originates with Melvin Goldstein who first reported about it in 1967. It is of some importance in academic research. Using MO theory the bicyclo[3.2.2]nonatrienyl cation was predicted to be destabilised and the corresponding anion predicted to be stabilised by bicycloaromaticity.