
In vertebrates, most neuronal cell axons are encased in myelin. Simply put, myelin insulates axons and increases the rate at which electrical impulses are passed along the axon. The myelinated axon can be likened to an electrical wire with insulating material (myelin) around it. However, unlike the plastic covering on an electrical wire, myelin does not form a single long sheath over the entire length of the axon. Rather, myelin ensheaths the axon in segments: in general, each axon is encased in multiple long myelin sheaths separated by short gaps called nodes of Ranvier.

Pelizaeus–Merzbacher disease is an X-linked neurological disorder that damages oligodendrocytes in the central nervous system. It is caused by mutations in proteolipid protein 1 (PLP1), a major myelin protein. It is characterized by a decrease in the amount of insulating myelin surrounding the nerves (hypomyelination) and belongs to a group of genetic diseases referred to as leukodystrophies.
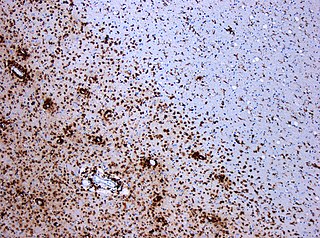
A demyelinating disease refers to any disease affecting the nervous system where the myelin sheath surrounding neurons is damaged. This damage disrupts the transmission of signals through the affected nerves, resulting in a decrease in their conduction ability. Consequently, this reduction in conduction can lead to deficiencies in sensation, movement, cognition, or other functions depending on the nerves affected.

Alexander disease is a very rare autosomal dominant leukodystrophy, which are neurological conditions caused by anomalies in the myelin which protects nerve fibers in the brain. The most common type is the infantile form that usually begins during the first two years of life. Symptoms include mental and physical developmental delays, followed by the loss of developmental milestones, an abnormal increase in head size and seizures. The juvenile form of Alexander disease has an onset between the ages of 2 and 13 years. These children may have excessive vomiting, difficulty swallowing and speaking, poor coordination, and loss of motor control. Adult-onset forms of Alexander disease are less common. The symptoms sometimes mimic those of Parkinson’s disease or multiple sclerosis, or may present primarily as a psychiatric disorder.

Leukodystrophies are a group of, usually, inherited disorders, characterized by degeneration of the white matter in the brain. The word leukodystrophy comes from the Greek roots leuko, "white", dys, "abnormal" and troph, "growth". The leukodystrophies are caused by imperfect growth or development of the glial cells which produce the myelin sheath, the fatty insulating covering around nerve fibers. Leukodystrophies may be classified as hypomyelinating or demyelinating diseases, respectively, depending on whether the damage is present before birth or occurs after. Other demyelinating diseases are usually not congenital and have a toxic or autoimmune cause.

Multiple sclerosis is an inflammatory demyelinating disease of the CNS in which activated immune cells invade the central nervous system and cause inflammation, neurodegeneration, and tissue damage. The underlying cause is currently unknown. Current research in neuropathology, neuroimmunology, neurobiology, and neuroimaging, together with clinical neurology, provide support for the notion that MS is not a single disease but rather a spectrum.
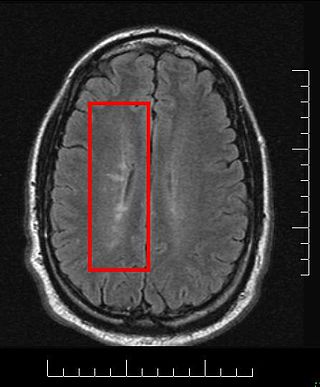
Multiple sclerosis and other demyelinating diseases of the central nervous system (CNS) produce lesions and glial scars or scleroses. They present different shapes and histological findings according to the underlying condition that produces them.

Translation initiation factor eIF-2B subunit epsilon is a protein that in humans is encoded by the EIF2B5 gene.

Membrane protein MLC1 is a protein that in humans is encoded by the MLC1 gene.

Translation initiation factor eIF-2B subunit beta is a protein that in humans is encoded by the EIF2B2 gene.

Translation initiation factor eIF-2B subunit alpha is a protein that in humans is encoded by the EIF2B1 gene.

Translation initiation factor eIF-2B subunit delta is a protein that in humans is encoded by the EIF2B4 gene.

Translation initiation factor eIF-2B subunit gamma is a protein that in humans is encoded by the EIF2B3 gene.
Leukoencephalopathy with neuroaxonal spheroids (LENAS) is an extremely rare kind of leukoencephalopathy and is classified as a neurodegenerative disease. LENAS is a cause of severe and subacute dementia that results from damage to certain areas of the brain. This damage is to a type of brain tissue called white matter and axon damage due to swellings which are termed spheroids.
Eukaryotic Initiation Factor 2 (eIF2) is an eukaryotic initiation factor. It is required for most forms of eukaryotic translation initiation. eIF2 mediates the binding of tRNAiMet to the ribosome in a GTP-dependent manner. eIF2 is a heterotrimer consisting of an alpha, a beta, and a gamma subunit.
Megalencephalic leukoencephalopathy with subcortical cysts is a form of hereditary CNS demyelinating disease. It belongs to a group of disorders called leukodystrophies. It is characterized by early-onset enlargement of the head (macrocephaly) as well as delayed-onset neurological deterioration to include spasticity, epilepsy, and lack of muscular coordination. MLC does not appear to be a disease that is fatal at birth or early in life despite its symptoms, although the number of patients throughout history known to have the disease is fairly limited.
Ribose-5-phosphate isomerase deficiency is a human disorder caused by mutations in ribose-5-phosphate isomerase, an enzyme of the pentose phosphate pathway. With only four diagnosed patients over a 27-year period, RPI deficiency is the second rarest disease known as of now, being beaten only by Fields Condition affecting three individuals, Catherine and Kirstie Fields, and one unknown person.

Hereditary diffuse leukoencephalopathy with spheroids (HDLS) is a rare adult onset autosomal dominant disorder characterized by cerebral white matter degeneration with demyelination and axonal spheroids leading to progressive cognitive and motor dysfunction. Spheroids are axonal swellings with discontinuous or absence of myelin sheaths. It is believed that the disease arises from primary microglial dysfunction that leads to secondary disruption of axonal integrity, neuroaxonal damage, and focal axonal spheroids leading to demyelination. Spheroids in HDLS resemble to some extent those produced by shear stress in a closed head injury with damage to axons, causing them to swell due to blockage of axoplasmic transport. In addition to trauma, axonal spheroids can be found in aged brain, stroke, and in other degenerative diseases. In HDLS, it is uncertain whether demyelination occurs prior to the axonal spheroids or what triggers neurodegeneration after apparently normal brain and white matter development, although genetic deficits suggest that demyelination and axonal pathology may be secondary to microglial dysfunction. The clinical syndrome in patients with HDLS is not specific and it can be mistaken for Alzheimer's disease, frontotemporal dementia, atypical Parkinsonism, multiple sclerosis, or corticobasal degeneration.

Hypomyelination-congenital cataract syndrome is a rare autosomal recessive hereditary disorder that affects the brain's white matter and is characterized by congenital cataract, psychomotor development delays, and moderate intellectual disabilities. It is a type of leukoencephalopathy.
Autosomal dominant leukodystrophy with autonomic disease is a rare neurological condition of genetic origin which is characterized by gradual demyelination of the central nervous system which results in various impairments, including ataxia, mild cognitive disability and autonomic dysfunction. It is part of a group of disorders called "leukodystrophies".

