Pancreatic cancer arises when cells in the pancreas, a glandular organ behind the stomach, begin to multiply out of control and form a mass. These cancerous cells have the ability to invade other parts of the body. A number of types of pancreatic cancer are known.

An insulinoma is a tumour of the pancreas that is derived from beta cells and secretes insulin. It is a rare form of a neuroendocrine tumour. Most insulinomas are benign in that they grow exclusively at their origin within the pancreas, but a minority metastasize. Insulinomas are one of the functional pancreatic neuroendocrine tumour (PNET) group. In the Medical Subject Headings classification, insulinoma is the only subtype of "islet cell adenoma".
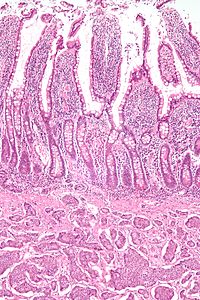
A carcinoid is a slow-growing type of neuroendocrine tumor originating in the cells of the neuroendocrine system. In some cases, metastasis may occur. Carcinoid tumors of the midgut are associated with carcinoid syndrome.
A VIPoma or vipoma is a rare endocrine tumor that overproduces vasoactive intestinal peptide. The incidence is about 1 per 10,000,000 per year. VIPomas usually originate from the non-β islet cells of the pancreas. They are sometimes associated with multiple endocrine neoplasia type 1. Roughly 50–75% of VIPomas are malignant, but even when they are benign, they are problematic because they tend to cause a specific syndrome: the massive amounts of VIP cause a syndrome of profound and chronic watery diarrhea and resultant dehydration, hypokalemia, achlorhydria, acidosis, flushing and hypotension, hypercalcemia, and hyperglycemia. This syndrome is called Verner–Morrison syndrome (VMS), WDHA syndrome, or pancreatic cholera syndrome (PCS). The eponym reflects the physicians who first described the syndrome.

Gastrinomas are neuroendocrine tumors (NETs), usually located in the duodenum or pancreas, that secrete gastrin and cause a clinical syndrome known as Zollinger–Ellison syndrome (ZES). A large number of gastrinomas develop in the pancreas or duodenum, with near-equal frequency, and approximately 10% arise as primary neoplasms in lymph nodes of the pancreaticoduodenal region.
Somatostatinomas are a tumor of the delta cells of the endocrine pancreas that produces somatostatin. Increased levels of somatostatin inhibit pancreatic hormones and gastrointestinal hormones. Thus, somatostatinomas are associated with mild diabetes mellitus, steatorrhoea and gallstones, and achlorhydria. Somatostatinomas are commonly found in the head of pancreas. Only ten percent of somatostatinomas are functional tumours [9], and 60–70% of tumours are malignant. Nearly two-thirds of patients with malignant somatostatinomas will present with metastatic disease.
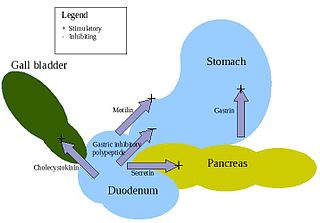
Enteroendocrine cells are specialized cells of the gastrointestinal tract and pancreas with endocrine function. They produce gastrointestinal hormones or peptides in response to various stimuli and release them into the bloodstream for systemic effect, diffuse them as local messengers, or transmit them to the enteric nervous system to activate nervous responses. Enteroendocrine cells of the intestine are the most numerous endocrine cells of the body. They constitute an enteric endocrine system as a subset of the endocrine system just as the enteric nervous system is a subset of the nervous system. In a sense they are known to act as chemoreceptors, initiating digestive actions and detecting harmful substances and initiating protective responses. Enteroendocrine cells are located in the stomach, in the intestine and in the pancreas. Microbiota play key roles in the intestinal immune and metabolic responses in these enteroendocrine cells via their fermentation product, acetate.
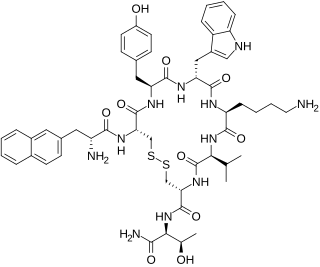
Lanreotide, sold under the brand name Somatuline among others, is a medication used in the management of acromegaly and symptoms caused by neuroendocrine tumors, most notably carcinoid syndrome. It is a long-acting analogue of somatostatin, like octreotide.

Somatostatin receptor type 2 is a protein that in humans is encoded by the SSTR2 gene.
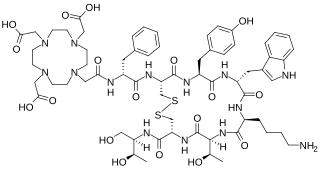
Edotreotide (USAN, also known as (DOTA0-Phe1-Tyr3) octreotide, DOTA-TOC, DOTATOC) is a substance which, when bound to various radionuclides, is used in the treatment and diagnosis of certain types of cancer. When used therapeutically it is an example of peptide receptor radionuclide therapy.

A pancreatic tumor is an abnormal growth in the pancreas. In adults, almost 90% are pancreatic cancer and a few are benign. Pancreatic tumors are rare in children.
Advanced Accelerator Applications is a France-based pharmaceutical group, specialized in the field of nuclear medicine. The group operates in all three segments of nuclear medicine to diagnose and treat serious conditions in the fields of oncology, neurology, cardiology, infectious and inflammatory diseases.
Pulmonary neuroendocrine tumors are neuroendocrine tumors localized to the lung: bronchus or pulmonary parenchyma.
Neuroendocrine differentiation is a term primarily used in relation to prostate cancers that display a significant neuroendocrine cell population on histopathological examination. These types of prostate cancer comprise true neuroendocrine cancers, such as small cell carcinoma, carcinoid and carcinoid-like tumors, as well as prostatic adenocarcinoma exhibiting focal neuroendocrine phenotype.
Pancreatic neuroendocrine tumours, often referred to as "islet cell tumours", or "pancreatic endocrine tumours" are neuroendocrine neoplasms that arise from cells of the endocrine (hormonal) and nervous system within the pancreas.
Hepatic artery embolization, also known as trans-arterial embolization (TAE), is one of the several therapeutic methods to treat primary liver tumors or metastases to the liver. The embolization therapy can reduce the size of the tumor, and decrease the tumor's impact such its hormone production, effectively decreasing symptoms. The treatment was initially developed in the early 1970s. The several types of hepatic artery treatments are based on the observation that tumor cells get nearly all their nutrients from the hepatic artery, while the normal cells of the liver get about 70-80 percent of their nutrients and 50% their oxygen supply from the portal vein, and thus can survive with the hepatic artery effectively blocked. In practice, hepatic artery embolization occludes the blood flow to the tumors, achieving significant tumor shrinkage in over 80% of people. Shrinkage rates vary.

Peptide receptor radionuclide therapy (PRRT) is a type of radionuclide therapy, using a radiopharmaceutical that targets peptide receptors to deliver localised treatment, typically for neuroendocrine tumours (NETs).

Lutetium (177Lu) oxodotreotide (INN) or 177Lu DOTA-TATE, trade name Lutathera, is a chelated complex of a radioisotope of the element lutetium with DOTA-TATE, used in peptide receptor radionuclide therapy (PRRT). Specifically, it is used in the treatment of cancers which express somatostatin receptors.

Somatostatin receptor antagonists are a class of chemical compounds that work by imitating the structure of the neuropeptide somatostatin. The somatostatin receptors are G protein-coupled receptors. Somatostatin receptor subtypes in humans are sstr1, 2A, 2 B, 3, 4 and 5. While normally expressed in the gastrointestinal (GI) tract, pancreas, hypothalamus, and central nervous system (CNS), they are expressed in different types of tumours. The predominant subtype in cancer cells is the ssrt2 subtype, which is expressed in neuroblastomas, meningiomas, medulloblastomas, breast carcinomas, lymphomas, renal cell carcinomas, paragangliomas, small cell lung carcinomas and hepatocellular carcinomas.

Somatostatin receptor antagonists are a class of chemical compounds that work by imitating the structure of the neuropeptide somatostatin, which is an endogenous hormone found in the human body. The somatostatin receptors are G protein-coupled receptors. Somatostatin receptor subtypes in humans include sstr1, 2A, 2 B, 3, 4, and 5. While normally expressed in the gastrointestinal (GI) tract, pancreas, hypothalamus, and central nervous system (CNS), they are expressed in different types of tumours. The predominant subtype in cancer cells is the ssrt2 subtype, which is expressed in neuroblastomas, meningiomas, medulloblastomas, breast carcinomas, lymphomas, renal cell carcinomas, paragangliomas, small cell lung carcinomas, and hepatocellular carcinomas.