Related Research Articles

Familial Mediterranean fever (FMF) is a hereditary inflammatory disorder. FMF is an autoinflammatory disease caused by mutations in Mediterranean fever gene, which encodes a 781–amino acid protein called pyrin. While all ethnic groups are susceptible to FMF, it usually occurs in people of Mediterranean origin—including Sephardic Jews, Mizrahi Jews, Ashkenazi Jews, Assyrians, Armenians, Azerbaijanis, Druze, Levantines, Kurds, Greeks, Turks and Italians.
Congenital insensitivity to pain (CIP), also known as congenital analgesia, is one or more extraordinarily rare conditions in which a person cannot feel physical pain. The conditions described here are separate from the HSAN group of disorders, which have more specific signs and cause. Because feeling physical pain is vital for survival, CIP is an extremely dangerous condition. It is common for people with the condition to die in childhood due to injuries or illnesses going unnoticed. Burn injuries are among the more common injuries.

Erythromelalgia or Mitchell's disease is a rare vascular peripheral pain disorder in which blood vessels, usually in the lower extremities or hands, are episodically blocked, then become hyperemic and inflamed. There is severe burning pain and skin redness. The attacks are periodic and are commonly triggered by heat, pressure, mild activity, exertion, insomnia or stress. Erythromelalgia may occur either as a primary or secondary disorder. Secondary erythromelalgia can result from small fiber peripheral neuropathy of any cause, polycythemia vera, essential thrombocytosis, hypercholesterolemia, mushroom or mercury poisoning, and some autoimmune disorders. Primary erythromelalgia is caused by mutation of the voltage-gated sodium channel α-subunit gene SCN9A.
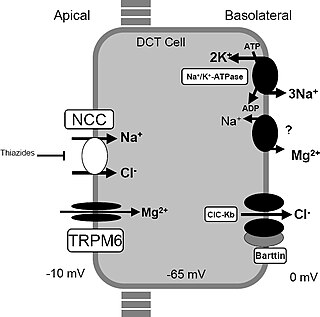
Gitelman syndrome (GS) is an autosomal recessive kidney tubule disorder characterized by low blood levels of potassium and magnesium, decreased excretion of calcium in the urine, and elevated blood pH. It is the most frequent hereditary salt-losing tubulopathy. Gitelman syndrome is caused by disease-causing variants on both alleles of the SLC12A3 gene. The SLC12A3 gene encodes the thiazide-sensitive sodium-chloride cotransporter, which can be found in the distal convoluted tubule of the kidney.
Myotonia congenita is a congenital neuromuscular channelopathy that affects skeletal muscles. It is a genetic disorder. The hallmark of the disease is the failure of initiated contraction to terminate, often referred to as delayed relaxation of the muscles (myotonia) and rigidity. Symptoms include delayed relaxation of the muscles after voluntary contraction (myotonia), and may also include stiffness, hypertrophy (enlargement), transient weakness in some forms of the disorder, severe masseter spasm, and cramping. The condition is sometimes referred to as fainting goat syndrome, as it is responsible for the eponymous 'fainting' seen in fainting goats when presented with a sudden stimulus. Of note, myotonia congenita has no association with malignant hyperthermia (MH).
Periodic paralysis is a group of rare genetic diseases that lead to weakness or paralysis from common triggers such as cold, heat, high carbohydrate meals, not eating, stress or excitement and physical activity of any kind. The underlying mechanism of these diseases are malfunctions in the ion channels in skeletal muscle cell membranes that allow electrically charged ions to leak in or out of the muscle cell, causing the cell to depolarize and become unable to move.

Paramyotonia congenita (PC) is a rare congenital autosomal dominant neuromuscular disorder characterized by "paradoxical" myotonia. This type of myotonia has been termed paradoxical because it becomes worse with exercise whereas classical myotonia, as seen in myotonia congenita, is alleviated by exercise. PC is also distinguished as it can be induced by cold temperatures. Although more typical of the periodic paralytic disorders, patients with PC may also have potassium-provoked paralysis. PC typically presents within the first decade of life and has 100% penetrance. Patients with this disorder commonly present with myotonia in the face or upper extremities. The lower extremities are generally less affected. While some other related disorders result in muscle atrophy, this is not normally the case with PC. This disease can also present as hyperkalemic periodic paralysis and there is debate as to whether the two disorders are actually distinct.
Familial hemiplegic migraine (FHM) is an autosomal dominant type of hemiplegic migraine that typically includes weakness of half the body which can last for hours, days, or weeks. It can be accompanied by other symptoms, such as ataxia, coma, and paralysis. Migraine attacks may be provoked by minor head trauma. Some cases of minor head trauma in patients with hemiplegic migraine can develop into delayed cerebral edema, a life-threatening medical emergency. Clinical overlap occurs in some FHM patients with episodic ataxia type 2 and spinocerebellar ataxia type 6, benign familial infantile epilepsy, and alternating hemiplegia of childhood.
Alternating hemiplegia of childhood (AHC) is an ultra-rare neurological disorder named for the transient episodes, often referred to as "attacks", of hemiplegia that those with the condition experience. It typically presents before the age of 18 months. These hemiplegic attacks can cause anything from mild weakness to complete paralysis on one or both sides of the body, and they can vary greatly in duration. Attacks may also alternate from one side of the body to the other, or alternate between affecting one or both sides during a single attack. Besides hemiplegia, symptoms of the disorder include an extremely broad range of neurological and developmental impairments which are not well understood. Normally, hemiplegia and other associated symptoms cease completely with sleep, but they may recur upon waking.
Small fiber peripheral neuropathy is a type of peripheral neuropathy that occurs from damage to the small unmyelinated and myelinated peripheral nerve fibers. These fibers, categorized as C fibers and small Aδ fibers, are present in skin, peripheral nerves, and organs. The role of these nerves is to innervate some skin sensations and help control autonomic function. It is estimated that 15–20 million people in the United States have some form of peripheral neuropathy.
Episodic ataxia (EA) is an autosomal dominant disorder characterized by sporadic bouts of ataxia with or without myokymia. There are seven types recognized but the majority are due to two recognized entities. Ataxia can be provoked by psychological stress or startle, or heavy exertion, including exercise. Symptoms can first appear in infancy. There are at least six loci for EA, of which 4 are known genes. Some patients with EA also have migraine or progressive cerebellar degenerative disorders, symptomatic of either familial hemiplegic migraine or spinocerebellar ataxia. Some patients respond to acetazolamide though others do not.

Nav1.7 is a sodium ion channel that in humans is encoded by the SCN9A gene. It is usually expressed at high levels in two types of neurons: the nociceptive (pain) neurons at dorsal root ganglion (DRG) and trigeminal ganglion and sympathetic ganglion neurons, which are part of the autonomic (involuntary) nervous system.
The paroxysmal dyskinesias (PD) are a group of movement disorders characterized by attacks of hyperkinesia with intact consciousness. Paroxysmal dyskinesia is a rare disorder, however the number of individuals it affects remains unclear. There are three different subtypes of PD that include paroxysmal kinesigenic dyskinesia (PKD), paroxysmal nonkinesigenic dyskinesia (PNKD), and paroxysmal exercise-induced dyskinesia (PED). Other neurological diseases have similar symptoms to PD, such as epilepsy and Parkinson's. The different subtypes make accurate and quick diagnosis of PD challenging. Thus, PD is often under reported and misdiagnosed, making it difficult to accurately study its prevalence in human populations. Onset of PD is usually in late childhood to early adolescence. New drug regimens help treat symptoms of PD, but no cure for the disorder is known.
Paroxysmal kinesigenic choreoathetosis (PKC) also called paroxysmal kinesigenic dyskinesia (PKD) is a hyperkinetic movement disorder characterized by attacks of involuntary movements, which are triggered by sudden voluntary movements. The number of attacks can increase during puberty and decrease in a person's 20s to 30s. Involuntary movements can take many forms such as ballism, chorea or dystonia and usually only affect one side of the body or one limb in particular. This rare disorder only affects about 1 in 150,000 people, with PKD accounting for 86.8% of all the types of paroxysmal dyskinesias, and occurs more often in males than females. There are two types of PKD, primary and secondary. Primary PKD can be further broken down into familial and sporadic. Familial PKD, which means the individual has a family history of the disorder, is more common, but sporadic cases are also seen. Secondary PKD can be caused by many other medical conditions such as multiple sclerosis (MS), stroke, pseudohypoparathyroidism, hypocalcemia, hypoglycemia, hyperglycemia, central nervous system trauma, or peripheral nervous system trauma. PKD has also been linked with infantile convulsions and choreoathetosis (ICCA) syndrome, in which patients have afebrile seizures during infancy and then develop paroxysmal choreoathetosis later in life. This phenomenon is actually quite common, with about 42% of individuals with PKD reporting a history of afebrile seizures as a child.
Vestibular migraine (VM) is vertigo with migraine, either as a symptom of migraine or as a related neurological disorder.
EAST syndrome is a syndrome consisting of epilepsy, ataxia, sensorineural deafness and salt-wasting renal tubulopathy. The tubulopathy in this condition predispose to hypokalemic metabolic alkalosis with normal blood pressure. Hypomagnesemia may also be present.

Vestibulocerebellar syndrome, also known as vestibulocerebellar ataxia, is a progressive neurological disorder that causes a variety of medical problems. Initially symptoms present as periodic attacks of abnormal eye movements but may intensify to longer-lasting motor incapacity. The disorder has been localized to the vestibulocerebellum, specifically the flocculonodular lobe. Symptoms of vestibulocerebellar syndrome may appear in early childhood but the full onset of neurological symptoms including nystagmus, ataxia, and tinnitus does not occur until early adulthood. To date, vestibulocerebellar syndrome has only been identified in three families but has affected multiple generations within them. Based on the familial pedigrees it has been characterized as an autosomal dominant disorder, although the exact genetic locus has not been identified. It has been found to be genetically distinct from other seemingly similar forms of neurological syndromes such as episodic ataxia types 1 and 2. Due to its rarity, however, little is known about specific details of the pathology or long-term treatment options. There is currently no cure for vestibulocerebellar syndrome, although some drug therapies have been effective in alleviating particular symptoms of the disorder.

Thyrotoxic periodic paralysis (TPP) is a rare condition featuring attacks of muscle weakness in the presence of hyperthyroidism. Hypokalemia is usually present during attacks. The condition may be life-threatening if weakness of the breathing muscles leads to respiratory failure, or if the low potassium levels lead to abnormal heart rhythms. If untreated, it is typically recurrent in nature.
Alternating hemiplegia is a form of hemiplegia that has an ipsilateral cranial nerve palsies and contralateral hemiplegia or hemiparesis of extremities of the body. The disorder is characterized by recurrent episodes of paralysis on one side of the body. There are multiple forms of alternating hemiplegia, Weber's syndrome, middle alternating hemiplegia, and inferior alternating hemiplegia. This type of syndrome can result from a unilateral lesion in the brainstem affecting both upper motor neurons and lower motor neurons. The muscles that would receive signals from these damaged upper motor neurons result in spastic paralysis. With a lesion in the brainstem, this affects the majority of limb and trunk muscles on the contralateral side due to the upper motor neurons decussation after the brainstem. The cranial nerves and cranial nerve nuclei are also located in the brainstem making them susceptible to damage from a brainstem lesion. Cranial nerves III (Oculomotor), VI (Abducens), and XII (Hypoglossal) are most often associated with this syndrome given their close proximity with the pyramidal tract, the location which upper motor neurons are in on their way to the spinal cord. Damages to these structures produce the ipsilateral presentation of paralysis or palsy due to the lack of cranial nerve decussation before innervating their target muscles. The paralysis may be brief or it may last for several days, many times the episodes will resolve after sleep. Some common symptoms of alternating hemiplegia are mental impairment, gait and balance difficulties, excessive sweating and changes in body temperature.
Louis Ptáček is an American neurologist and professor who contributed greatly to the field of genetics and neuroscience. He was also an HHMI investigator from 1997 to 2018. His chief areas of research include the understanding of inherited Mendelian disorders and circadian rhythm genes. Currently, Ptáček is a neurology professor and a director of the Division of Neurogenetics in University of California, San Francisco, School of Medicine. His current investigations primarily focus on extensive clinical studies in families with hereditary disorders, which include identifying and characterizing the genes responsible for neurological variations.
References
- 1 2 3 4 5 6 7 8 Fertleman CR, Ferrie CD, Aicardi J, et al. (2007). "Paroxysmal extreme pain disorder, (previously familial rectal pain syndrome)". Neurology. 69 (6): 586–595. doi:10.1212/01.wnl.0000268065.16865.5f. PMID 17679678. S2CID 20009430.
- 1 2 3 4 Fertleman CR, Baker MD, Parker KA, et al. (2006). "SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes". Neuron. 52 (5): 767–774. doi: 10.1016/j.neuron.2006.10.006 . PMID 17145499. S2CID 11715780.
- ↑ Elmslie FV, Wilson J, Rossiter MA (1996). "Familial rectal pain: is it under-diagnosed?". Journal of the Royal Society of Medicine. 89 (5): 290P–1P. doi:10.1177/014107689608900525. PMC 1295795 . PMID 8778439.
- ↑ HAYDEN R, GROSSMAN M (1959). "Rectal, ocular, and submaxillary pain; a familial autonomic disorder related to proctalgia fugaz: report of a family". AMA Journal of Diseases of Children. 97 (4): 479–82. doi:10.1001/archpedi.1959.02070010481013. PMID 13636703.
- 1 2 Fertleman CR, Ferrie CD (2006). "What's in a name--familial rectal pain syndrome becomes paroxysmal extreme pain disorder". J. Neurol. Neurosurg. Psychiatry. 77 (11): 1294–1295. doi:10.1136/jnnp.2006.089664. PMC 2077381 . PMID 17043302.
- ↑ Dugan RE (1972). "Familial rectal pain". Lancet. 1 (7755): 854. doi:10.1016/S0140-6736(72)90847-1. PMID 4111621.