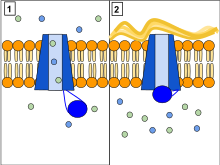
Ion channels are pore-forming membrane proteins that allow ions to pass through the channel pore. Their functions include establishing a resting membrane potential, shaping action potentials and other electrical signals by gating the flow of ions across the cell membrane, controlling the flow of ions across secretory and epithelial cells, and regulating cell volume. Ion channels are present in the membranes of all cells. Ion channels are one of the two classes of ionophoric proteins, the other being ion transporters.

Cystic fibrosis (CF) is a rare genetic disorder that affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine. The hallmark feature of CF is the accumulation of thick mucus in different organs. Long-term issues include difficulty breathing and coughing up mucus as a result of frequent lung infections. Other signs and symptoms may include sinus infections, poor growth, fatty stool, clubbing of the fingers and toes, and infertility in most males. Different people may have different degrees of symptoms.

Chloride channels are a superfamily of poorly understood ion channels specific for chloride. These channels may conduct many different ions, but are named for chloride because its concentration in vivo is much higher than other anions. Several families of voltage-gated channels and ligand-gated channels have been characterized in humans.
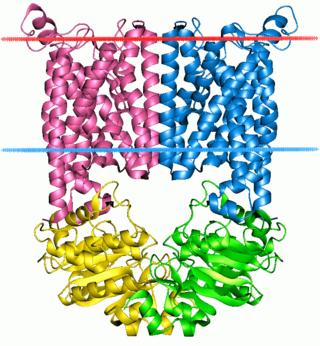
The ABC transporters, ATP synthase (ATP)-binding cassette transporters are a transport system superfamily that is one of the largest and possibly one of the oldest gene families. It is represented in all extant phyla, from prokaryotes to humans. ABC transporters belong to translocases.

The epithelial sodium channel(ENaC), (also known as amiloride-sensitive sodium channel) is a membrane-bound ion channel that is selectively permeable to sodium ions (Na+). It is assembled as a heterotrimer composed of three homologous subunits α or δ, β, and γ, These subunits are encoded by four genes: SCNN1A, SCNN1B, SCNN1G, and SCNN1D. The ENaC is involved primarily in the reabsorption of sodium ions at the collecting ducts of the kidney's nephrons. In addition to being implicated in diseases where fluid balance across epithelial membranes is perturbed, including pulmonary edema, cystic fibrosis, COPD and COVID-19, proteolyzed forms of ENaC function as the human salt taste receptor.

Arylsulfatase B is an enzyme associated with mucopolysaccharidosis VI.

Sodium-hydrogen antiporter 3 regulator 1 is a regulator of Sodium-hydrogen antiporter 3. It is encoded by the gene SLC9A3R1. It is also known as ERM Binding Protein 50 (EBP50) or Na+/H+ Exchanger Regulatory Factor (NHERF1). It is believed to interact via long-range allostery, involving significant protein dynamics.

MT-ND4 is a gene of the mitochondrial genome coding for the NADH-ubiquinone oxidoreductase chain 4 (ND4) protein. The ND4 protein is a subunit of NADH dehydrogenase (ubiquinone), which is located in the mitochondrial inner membrane and is the largest of the five complexes of the electron transport chain. Variations in the MT-ND4 gene are associated with age-related macular degeneration (AMD), Leber's hereditary optic neuropathy (LHON), mesial temporal lobe epilepsy (MTLE) and cystic fibrosis.

Multidrug resistance-associated protein 1 (MRP1) is a protein that in humans is encoded by the ABCC1 gene.

Heat shock protein HSP 90-beta also called HSP90beta is a protein that in humans is encoded by the HSP90AB1 gene.

Sodium-hydrogen exchange regulatory cofactor NHE-RF2 (NHERF-2) also known as tyrosine kinase activator protein 1 (TKA-1) or SRY-interacting protein 1 (SIP-1) is a protein that in humans is encoded by the SLC9A3R2 gene.

Golgi-associated PDZ and coiled-coil motif-containing protein is a protein that in humans is encoded by the GOPC gene.

A channel blocker is the biological mechanism in which a particular molecule is used to prevent the opening of ion channels in order to produce a physiological response in a cell. Channel blocking is conducted by different types of molecules, such as cations, anions, amino acids, and other chemicals. These blockers act as ion channel antagonists, preventing the response that is normally provided by the opening of the channel.

Pyocyanin (PCN−) is one of the many toxic compounds produced and secreted by the Gram negative bacterium Pseudomonas aeruginosa. Pyocyanin is a blue secondary metabolite, turning red below pH 4.9, with the ability to oxidise and reduce other molecules and therefore kill microbes competing against P. aeruginosa as well as mammalian cells of the lungs which P. aeruginosa has infected during cystic fibrosis. Since pyocyanin is a zwitterion at blood pH, it is easily able to cross the cell membrane. There are three different states in which pyocyanin can exist: oxidized (blue), monovalently reduced (colourless) or divalently reduced (red). Mitochondria play an important role in the cycling of pyocyanin between its redox states. Due to its redox-active properties, pyocyanin generates reactive oxygen species.
Transepithelial potential difference (TEPD) is the voltage across an epithelium, and is the sum of the membrane potentials for the outer and inner cell membranes.

The CFTR inhibitory factor (Cif) is a protein virulence factor secreted by the Gram-negative bacteria Pseudomonas aeruginosa and Acinetobacter nosocomialis. Discovered at Dartmouth Medical School, Cif is able to alter the trafficking of select ABC transporters in eukaryotic epithelial cells, such as the cystic fibrosis transmembrane conductance regulator (CFTR), and P-glycoprotein by interfering with the host deubiquitinating machinery. By promoting the ubiquitin-mediated degradation of CFTR, Cif is able to phenocopy cystic fibrosis at the cellular level. The cif gene is transcribed as part of a 3 gene operon, whose expression is negatively regulated by CifR, a TetR family repressor.

Ivacaftor is a medication used to treat cystic fibrosis in people with certain mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, who account for 4–5% cases of cystic fibrosis. It is also included in combination medications, lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor which are used to treat people with cystic fibrosis.
Chloride channel openers refer to a specific category of drugs which are involved in a wide variety of physiological functions and processes such as the regulation of neuroexcitation, transepithelial salt transport, and smooth muscle contraction. Due to their distribution throughout the body, diversity, functionality, and associated pathology, chloride channels represent an ideal target for the development of channel modulating drugs such as chloride channel openers.
Jue Chen is a Chinese-born American structural biologist and biochemist. She is the William E. Ford professor of biochemistry and head of the Laboratory of Membrane Biology and Biophysics at the Rockefeller University and a Howard Hughes Medical Institute investigator. Her research focuses on elucidating the structure and function of ATP-binding cassette (ABC) transporters.
Elexacaftor/tezacaftor/ivacaftor, sold under the brand names Trikafta and Kaftrio, is a fixed-dose combination medication used to treat cystic fibrosis. Elexacaftor/tezacaftor/ivacaftor is composed of a combination of ivacaftor, a chloride channel opener, and elexacaftor and tezacaftor, CFTR modulators.