
Tietz syndrome, also called Tietz albinism-deafness syndrome or albinism and deafness of Tietz, is an autosomal dominant congenital disorder characterized by deafness and leucism. It is caused by a mutation in the microphthalmia-associated transcription factor (MITF) gene. Tietz syndrome was first described in 1963 by Walter Tietz (1927–2003) a German Physician working in California.

Conductive hearing loss (CHL) occurs when there is a problem transferring sound waves anywhere along the pathway through the outer ear, tympanic membrane (eardrum), or middle ear (ossicles). If a conductive hearing loss occurs in conjunction with a sensorineural hearing loss, it is referred to as a mixed hearing loss. Depending upon the severity and nature of the conductive loss, this type of hearing impairment can often be treated with surgical intervention or pharmaceuticals to partially or, in some cases, fully restore hearing acuity to within normal range. However, cases of permanent or chronic conductive hearing loss may require other treatment modalities such as hearing aid devices to improve detection of sound and speech perception.

Sensorineural hearing loss (SNHL) is a type of hearing loss in which the root cause lies in the inner ear or sensory organ or the vestibulocochlear nerve. SNHL accounts for about 90% of reported hearing loss. SNHL is usually permanent and can be mild, moderate, severe, profound, or total. Various other descriptors can be used depending on the shape of the audiogram, such as high frequency, low frequency, U-shaped, notched, peaked, or flat.
Primrose syndrome is a rare, slowly progressive genetic disorder that can vary symptomatically between individual cases, but is generally characterised by ossification of the external ears, learning difficulties, and facial abnormalities. It was first described in 1982 in Scotland's Royal National Larbert Institution by Dr D.A.A. Primrose.
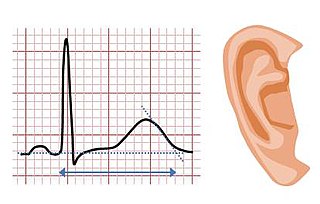
Jervell and Lange-Nielsen syndrome (JLNS) is a rare type of long QT syndrome associated with severe, bilateral sensorineural hearing loss. Those with JLNS are at risk of abnormal heart rhythms called arrhythmias, which can lead to fainting, seizures, or sudden death. JLNS, like other forms of long QT syndrome, causes the cardiac muscle to take longer than usual to recharge between beats. It is caused by genetic variants responsible for producing ion channels that carry transport potassium out of cells. The condition is usually diagnosed using an electrocardiogram, but genetic testing can also be used. Treatment includes lifestyle measures, beta blockers, and implantation of a defibrillator in some cases. It was first described by Anton Jervell and Fred Lange-Nielsen in 1957.
Mondini dysplasia, also known as Mondini malformation and Mondini defect, is an abnormality of the inner ear that is associated with sensorineural hearing loss.
Albinism-black lock-cell migration disorder is the initialism for the following terms and concepts that describe a condition affecting a person's physical appearance and physiology: (1) A – albinism, (2) B – black lock of hair, (3) C – cell migration disorder of the neurocytes of the gut, and (4) D – sensorineural deafness. The syndrome is caused by mutation in the endothelin B receptor gene (EDNRB).
Nonsyndromic deafness is hearing loss that is not associated with other signs and symptoms. In contrast, syndromic deafness involves hearing loss that occurs with abnormalities in other parts of the body. Genetic changes are related to the following types of nonsyndromic deafness.
Pendrin is an anion exchange protein that in humans is encoded by the SLC26A4 gene . Pendrin was initially identified as a sodium-independent chloride-iodide exchanger with subsequent studies showing that it also accepts formate and bicarbonate as substrates. Pendrin is similar to the Band 3 transport protein found in red blood cells. Pendrin is the protein which is mutated in Pendred syndrome, which is an autosomal recessive disorder characterized by sensorineural hearing loss, goiter and a partial organification problem detectable by a positive perchlorate test.

Thyroid dyshormonogenesis is a rare condition due to genetic defects in the synthesis of thyroid hormones.
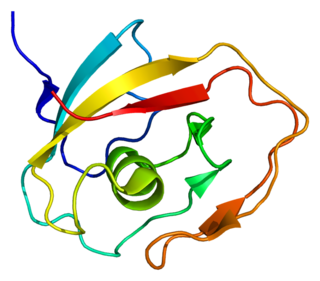
Cochlin is a protein that in humans is encoded by the COCH gene. It is an extracellular matrix (ECM) protein highly abundant in the cochlea and vestibule of the inner ear, constituting the major non-collagen component of the ECM of the inner ear. The protein is highly conserved in human, mouse, and chicken, showing 94% and 79% amino acid identity of human to mouse and chicken sequences, respectively.
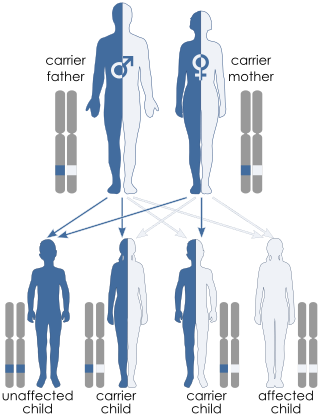
Björnstad syndrome is an autosomal recessive congenital condition involving pili torti, sensorineural deafness, and hair abnormalities. It was first characterized in 1965, in Oslo, by prof. Roar Theodor Bjørnstad after he observed an association between pili torti and hearing loss. The condition is extremely rare, with less than 50 cases documented in medical literature worldwide.
EAST syndrome is a syndrome consisting of epilepsy, ataxia, sensorineural deafness and salt-wasting renal tubulopathy. The tubulopathy in this condition predispose to hypokalemic metabolic alkalosis with normal blood pressure. Hypomagnesemia may also be present.

Large vestibular aqueduct is a structural deformity of the inner ear. Enlargement of this duct is one of the most common inner ear deformities and is commonly associated with hearing loss during childhood. The term was first discovered in 1791 by Mondini when he was completing a temporal bone dissection. It was then defined by Valvassori and Clemis as a vestibular aqueduct that is greater than or equal to 2.0 mm at the operculum and/or greater than or equal to 1.0 mm at the midpoint. Some use the term enlarged vestibular aqueduct syndrome, but this is felt by others to be erroneous as it is a clinical finding which can occur in several syndromes.

Mohr–Tranebjærg syndrome (MTS) is a rare X-linked recessive syndrome also known as deafness–dystonia syndrome and caused by mutation in the TIMM8A gene. It is characterized by clinical manifestations commencing with early childhood onset hearing loss, followed by adolescent onset progressive or , visual impairment from early adulthood onwards and dementia from the 4th decade onwards. The severity of the symptoms may vary, but they progress usually to severe deafness and dystonia and sometimes are accompanied by cortical deterioration of vision and mental deterioration.
Donnai–Barrow syndrome is a genetic disorder first described by Dian Donnai and Margaret Barrow in 1993. It is associated with LRP2. It is an inherited (genetic) disorder that affects many parts of the body.

Forkhead box I1 is a protein that in humans is encoded by the FOXI1 gene.
Nasodigitoacoustic syndrome, also called Keipert syndrome, is a rare congenital syndrome first described by J.A. Keipert and colleagues in 1973. The syndrome is characterized by a misshaped nose, broad thumbs and halluces, brachydactyly, sensorineural hearing loss, facial features such as hypertelorism, and developmental delay.

Distal 18q- is a genetic condition caused by a deletion of genetic material within one of the two copies of chromosome 18. The deletion involves the distal section of 18q and typically extends to the tip of the long arm of chromosome 18.
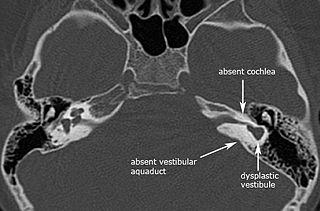
Michel aplasia, also known as complete labyrinthine aplasia (CLA), is a congenital abnormality of the inner ear. It is characterized by the bilateral absence of differentiated inner ear structures and results in complete deafness (anacusis). Michel aplasia should not be confused with michel dysplasia. It may affect one or both ears.

