
Marfan syndrome (MFS) is a multi-systemic genetic disorder that affects the connective tissue. Those with the condition tend to be tall and thin, with long arms, legs, fingers, and toes. They also typically have exceptionally flexible joints and abnormally curved spines. The most serious complications involve the heart and aorta, with an increased risk of mitral valve prolapse and aortic aneurysm. The lungs, eyes, bones, and the covering of the spinal cord are also commonly affected. The severity of the symptoms is variable.

Ehlers–Danlos syndromes (EDS) are a group of 13 genetic connective-tissue disorders in the current classification, with the latest type discovered in 2018. Symptoms often include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. These may be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis.

Tetralogy of Fallot (TOF), formerly known as Steno-Fallot tetralogy, is a congenital heart defect characterized by four specific cardiac defects. Classically, the four defects are:

MASA syndrome is a rare X-linked recessive neurological disorder on the L1 disorder spectrum belonging in the group of hereditary spastic paraplegias a paraplegia known to increase stiffness spasticity in the lower limbs. This syndrome also has two other names, CRASH syndrome and Gareis-Mason syndrome.

Takayasu's arteritis (TA), also known as aortic arch syndrome, nonspecific aortoarteritis, and pulseless disease, is a form of large vessel granulomatous vasculitis with massive intimal fibrosis and vascular narrowing, most commonly affecting young or middle-aged women of Asian descent, though anyone can be affected. It mainly affects the aorta and its branches, as well as the pulmonary arteries. Females are about 8–9 times more likely to be affected than males.

Oculocerebrorenal syndrome is a rare X-linked recessive disorder characterized by congenital cataracts, hypotonia, intellectual disability, proximal tubular acidosis, aminoaciduria and low-molecular-weight proteinuria. Lowe syndrome can be considered a cause of Fanconi syndrome.

Duane-radial ray syndrome, also known as Okihiro Syndrome, is a rare autosomal dominant disorder that primarily affects the eyes and causes abnormalities of bones in the arms and hands. This disorder is considered to be a SALL4-related disorder due to the SALL4 gene mutations leading to these abnormalities. It is diagnosed by clinical findings on a physical exam as well as genetic testing and imaging. After being diagnosed, there are other evaluations that one may go through in order to determine the extent of the disease. There are various treatments for the symptoms of this disorder.

Vertebral artery dissection (VAD) is a flap-like tear of the inner lining of the vertebral artery, which is located in the neck and supplies blood to the brain. After the tear, blood enters the arterial wall and forms a blood clot, thickening the artery wall and often impeding blood flow. The symptoms of vertebral artery dissection include head and neck pain and intermittent or permanent stroke symptoms such as difficulty speaking, impaired coordination, and visual loss. It is usually diagnosed with a contrast-enhanced CT or MRI scan.

Loeys–Dietz syndrome (LDS) is an autosomal dominant genetic connective tissue disorder. It has features similar to Marfan syndrome and Ehlers–Danlos syndrome. The disorder is marked by aneurysms in the aorta, often in children, and the aorta may also undergo sudden dissection in the weakened layers of the wall of the aorta. Aneurysms and dissections also can occur in arteries other than the aorta. Because aneurysms in children tend to rupture early, children are at greater risk for dying if the syndrome is not identified. Surgery to repair aortic aneurysms is essential for treatment.
Lymphedema–distichiasis syndrome is a medical condition associated with the FOXC2 gene. People with this hereditary condition have a double row of eyelashes, which is called distichiasis, and a risk of swollen limbs due to problems in the lymphatic system.

Kaufman oculocerebrofacial syndrome, also known as blepharophimosis-ptosis-intellectual disability syndrome, is an extremely rare autosomal recessive congenital disorder characterized by severe mental retardation, brachycephaly, upslanting palpebral fissures, eye abnormalities, and highly arched palate. It was characterized in 1971; eight cases had been identified as of 1995. To date, the amount of cases is disputed, with sources claiming the number ranges from 14 to 31.
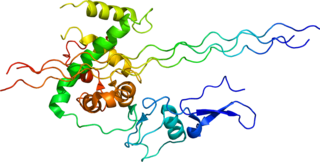
Type III Collagen is a homotrimer, or a protein composed of three identical peptide chains (monomers), each called an alpha 1 chain of type III collagen. Formally, the monomers are called collagen type III, alpha-1 chain and in humans are encoded by the COL3A1 gene. Type III collagen is one of the fibrillar collagens whose proteins have a long, inflexible, triple-helical domain.
Congenital contractural arachnodactyly (CCA), also known as Beals-Hecht syndrome, is a rare autosomal dominant congenital connective tissue disorder. As with Marfan syndrome, people with CCA typically have an arm span that is greater than their height and very long fingers and toes. However, Beals and Hecht discovered in 1972 that, unlike Marfan's, CCA is caused by mutations to the fibrillin-2 (FBN2) gene rather than the fibrillin-1 (FBN1) gene.
Buschke–Ollendorff syndrome (BOS) is a rare genetic skin disorder associated with LEMD3, that typically presents with widespread painless papules.

Solute carrier family 2, facilitated glucose transporter member 10 is a protein that in humans is encoded by the SLC2A10 gene.
Keutel syndrome (KS) is a rare autosomal recessive genetic disorder characterized by abnormal diffuse cartilage calcification, hypoplasia of the mid-face, peripheral pulmonary stenosis, hearing loss, short distal phalanges (tips) of the fingers and mild mental retardation. Individuals with KS often present with peripheral pulmonary stenosis, brachytelephalangism, sloping forehead, midface hypoplasia, and receding chin. It is associated with abnormalities in the gene coding for matrix gla protein, MGP. Being an autosomal recessive disorder, it may be inherited from two unaffected, abnormal MGP-carrying parents. Thus, people who inherit two affected MGP alleles will likely inherit KS.
Parkes Weber syndrome (PWS) is a congenital disorder of the vascular system. It is an extremely rare condition, and its exact prevalence is unknown. It is named after British dermatologist Frederick Parkes Weber, who first described the syndrome in 1907.

McKusick–Kaufman syndrome is a genetic condition associated with MKKS.
Cantú syndrome is a rare condition characterized by hypertrichosis, osteochondrodysplasia, and cardiomegaly. Less than 50 cases have been described in the literature; they are associated with a mutation in the ABCC9-gene that codes for the ABCC9-protein.

L1 syndrome is a group of mild to severe X-linked recessive disorders that share a common genetic basis. The spectrum of L1 syndrome disorders includes X-linked complicated corpus callosum dysgenesis, spastic paraplegia 1, MASA syndrome, and X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS). It is also called L1CAM syndrome and CRASH syndrome, an acronym for its primary clinical features: corpus callosum hypoplasia, retardation, adducted thumbs, spasticity, and hydrocephalus.


